Genetic and Antigenic Evolution of European Swine Influenza A Viruses of HA-1C (Avian-Like) and HA-1B (Human-Like) Lineages in France from 2000 to 2018

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Samples, swIAV Detection and Preliminary Molecular Subtyping
2.2. Virus Isolation
2.3. Sequencing
2.4. Phylogenetic Analyses of HA-, NA- and M-Encoding Genes
2.5. Large-Scale Phylogeny of M-Encoding Segment
2.6. Amino Acid Sequence Analyses
2.7. Antigenic Characterization
3. Results
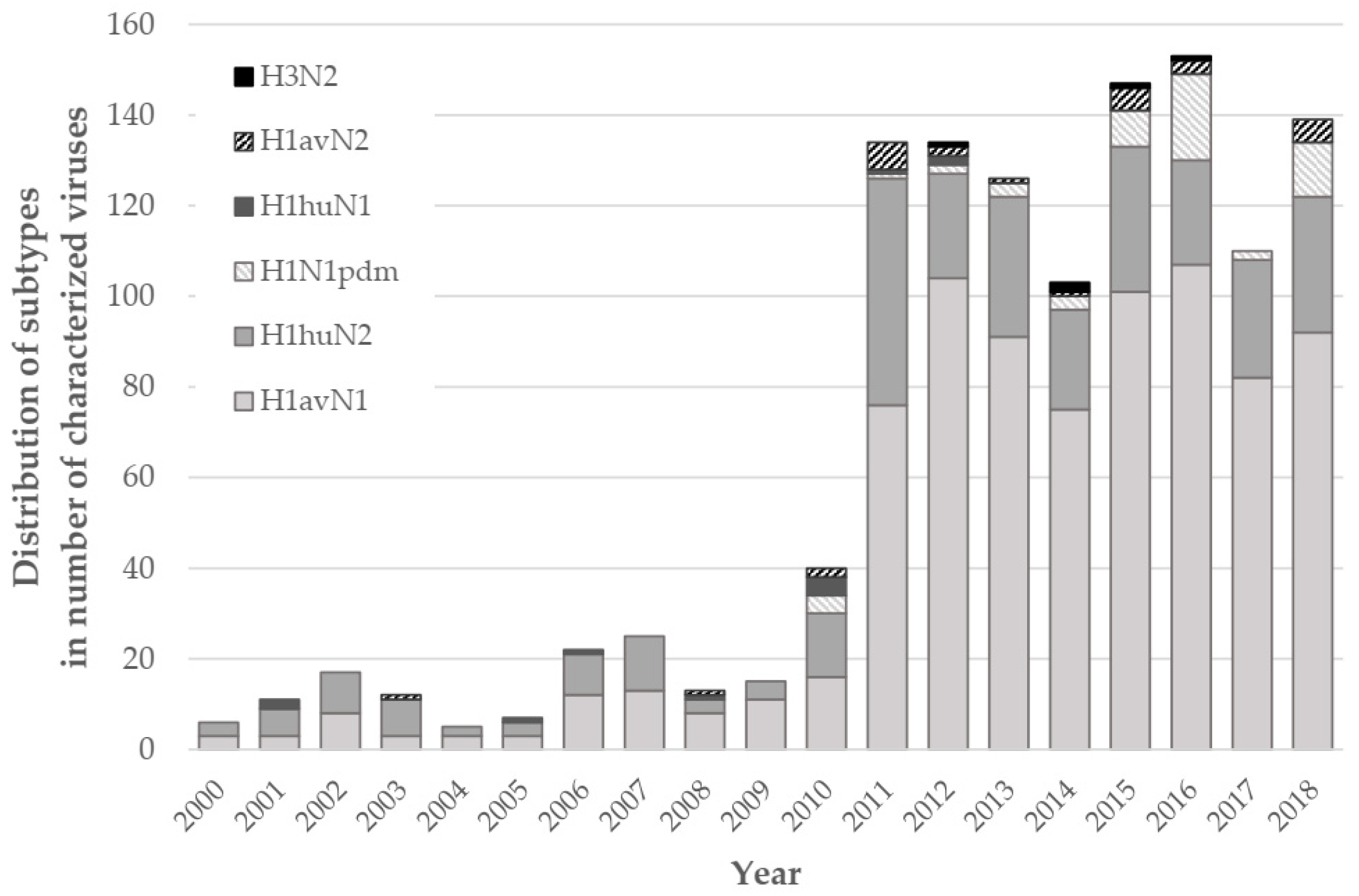
3.1. Relative Proportions of H1avNy and H1huNy swIAVs in France between 2000 and 2018
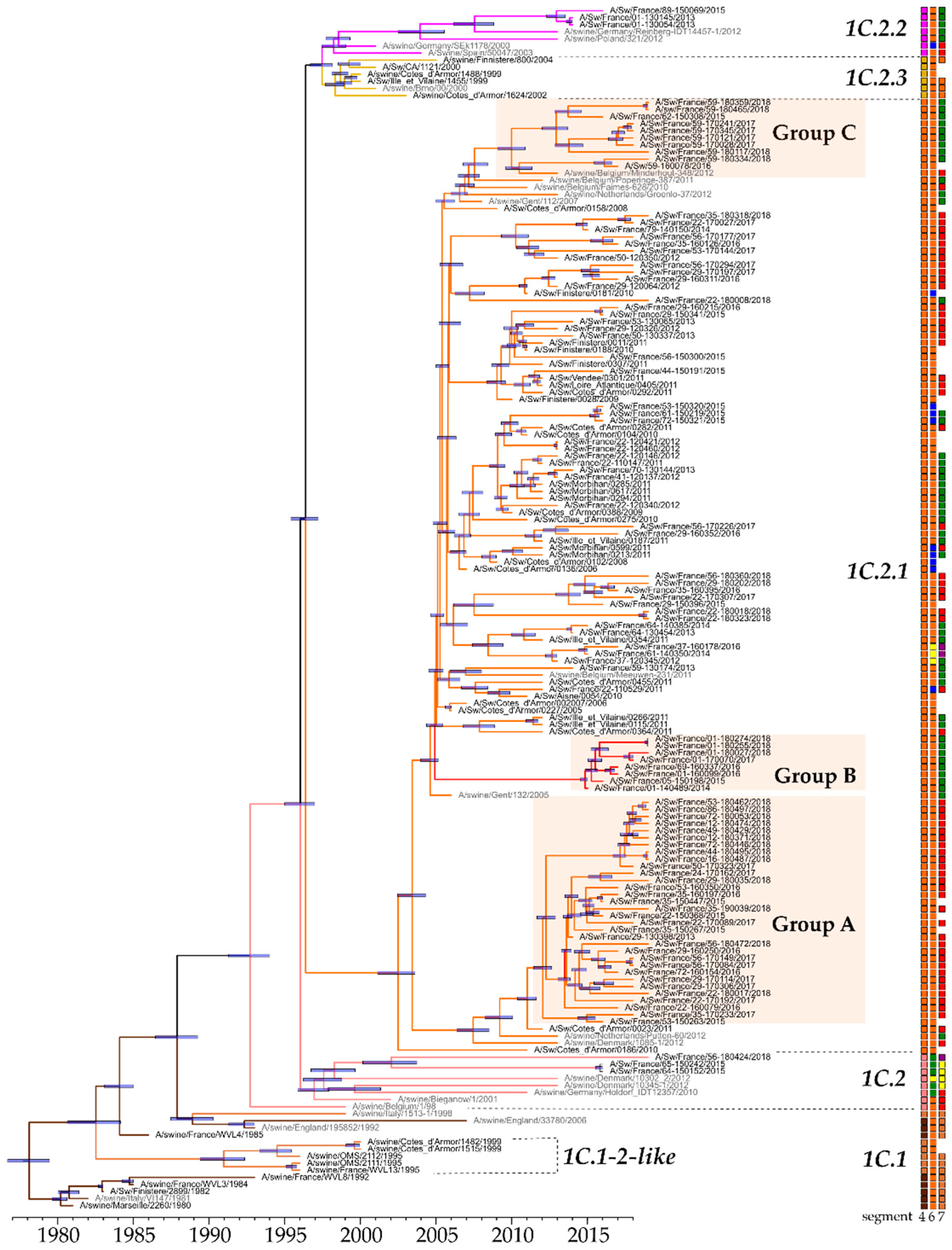
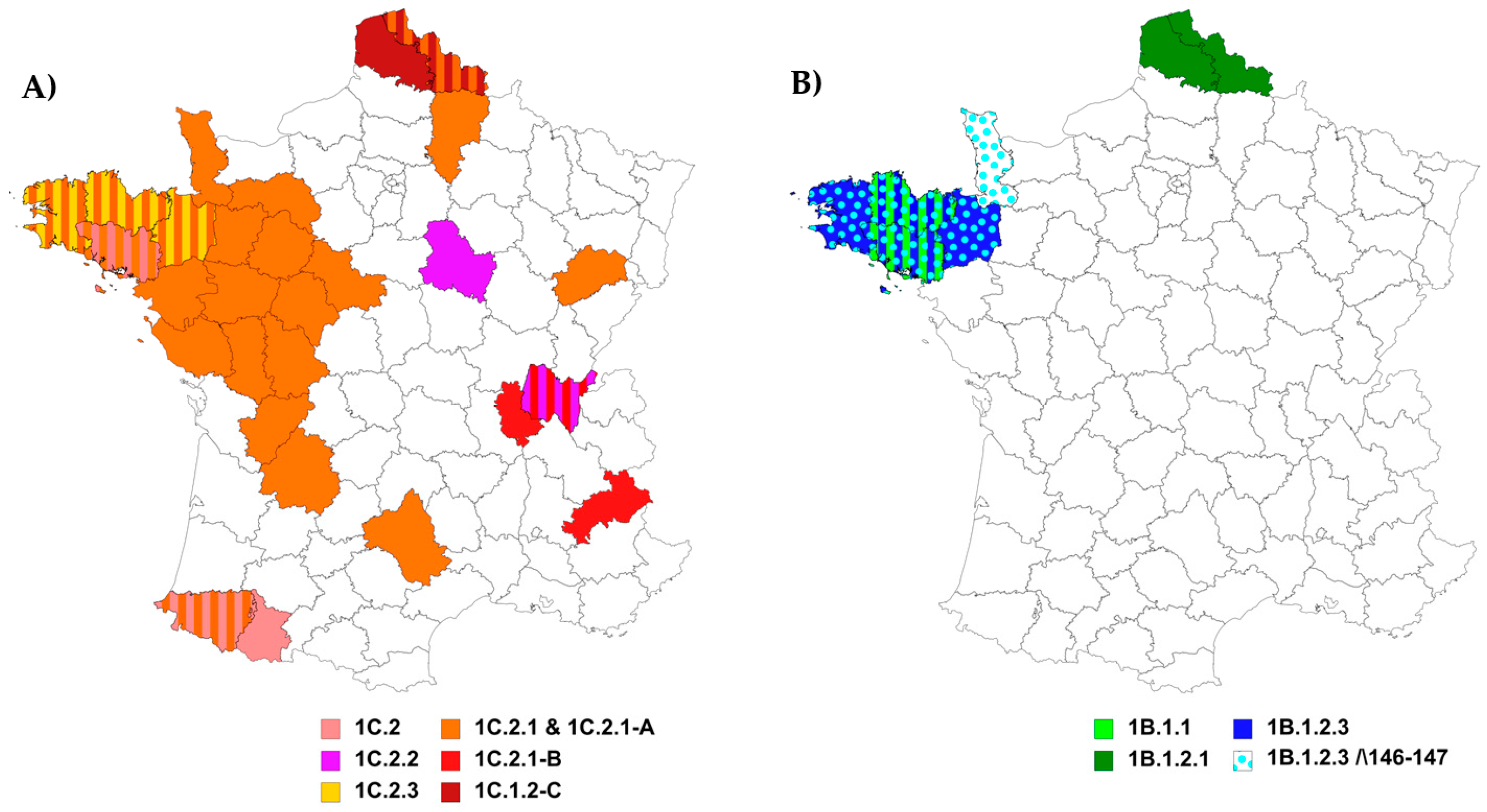
3.2. Genetic and Antigenic Evolution of HA-1C (H1avNy) swIAVs Isolated in France
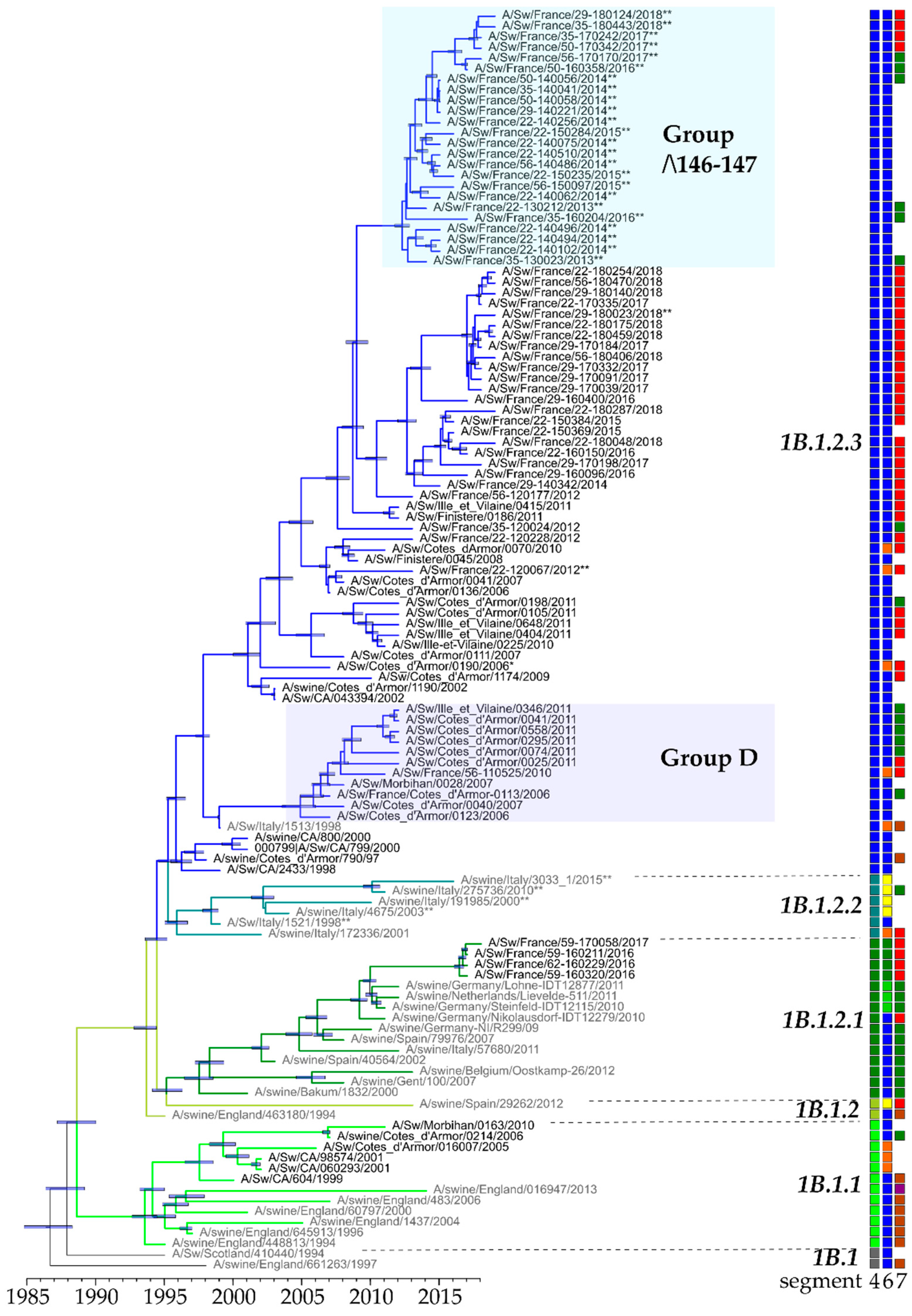
3.3. Genetic and Antigenic Evolution of HA-1B (H1huNy) swIAVs Isolated in France
3.4. Genetic Evolution of NA-, M- and other Protein-Encoding Genes from H1av (HA-1C) and H1hu (HA-1B) swIAVs
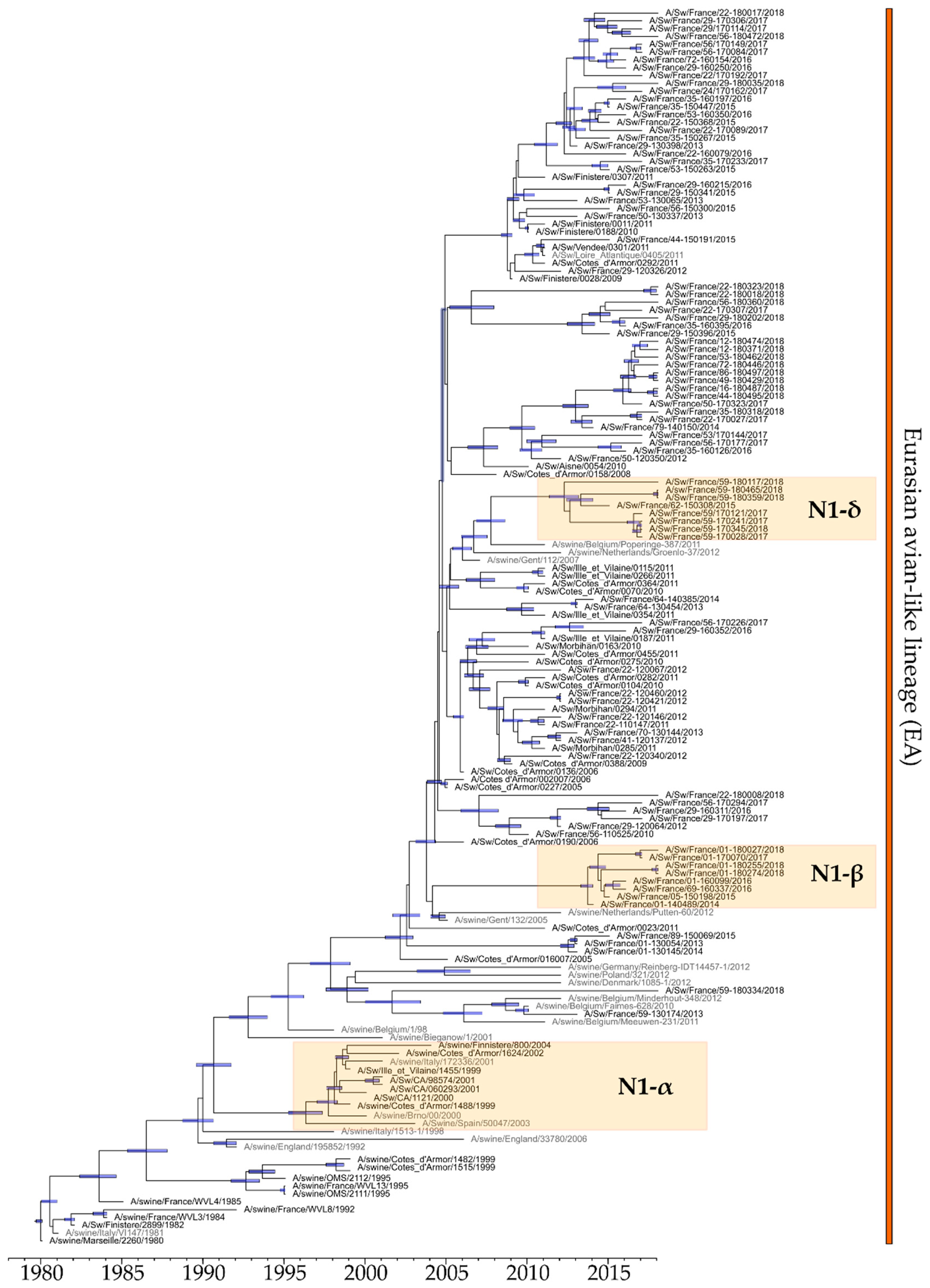
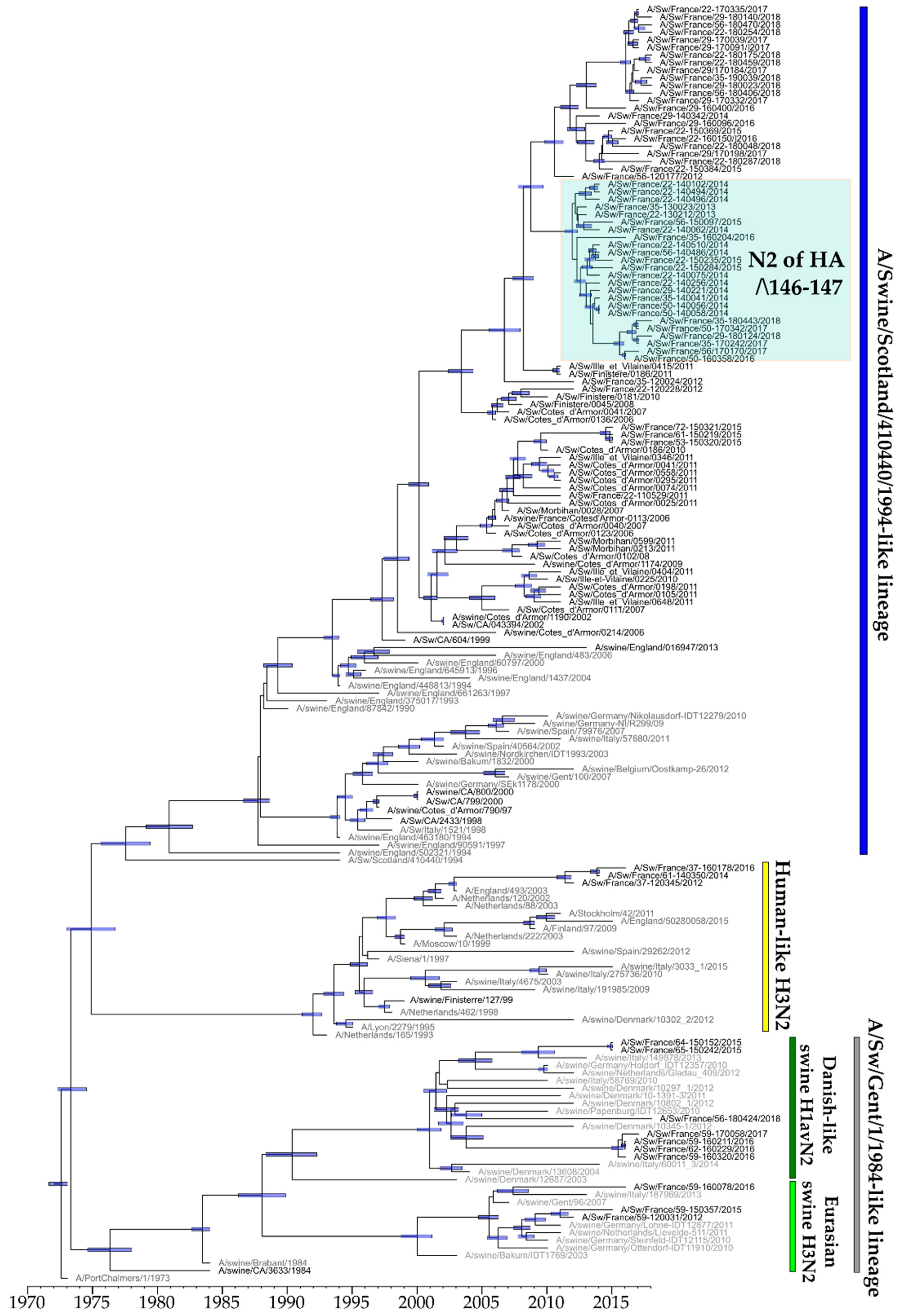
3.4.1. Evolution of NA-Encoding Segments of N1 and N2 Subtypes
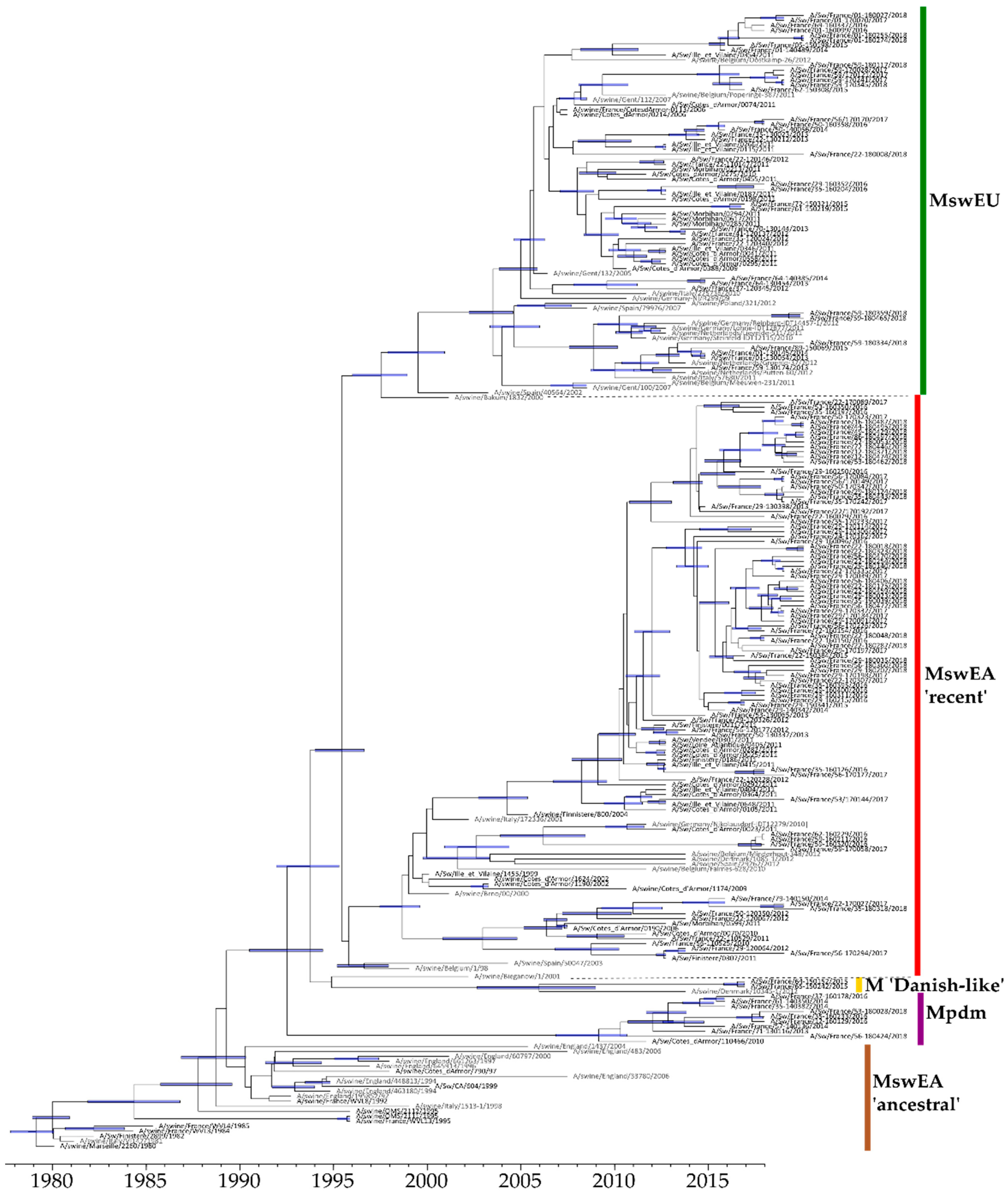
3.4.2. Evolution of M-Encoding Segment
3.4.3. Evolution of Other Internal Protein-Encoding Genomic Segments
3.5. Overview of H1av and H1hu Genotypes Identified in France from 2000 to 2018
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Webster, R.G.; Laver, W.G.; Air, G.M.; Schild, G.C. Molecular mechanisms of variation in influenza viruses. Nature 1982, 296, 115. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Richard, M.; Verhagen, J.H.; van Riel, D.; Schrauwen, E.J.A.; van den Brand, J.M.A.; Mänz, B.; Bodewes, R.; Herfst, S. One health, multiple challenges: The inter-species transmission of influenza A virus. One Health 2015, 1, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tscherne, D.M.; García-Sastre, A. Virulence determinants of pandemic influenza viruses. J. Clin. Investig. 2011, 121, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Carrat, F.; Flahault, A. Influenza vaccine: The challenge of antigenic drift. Vaccine 2007, 25, 6852–6862. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, N.M.; Galvani, A.P.; Bush, R.M. Ecological and immunological determinants of influenza evolution. Nature 2003, 422, 428. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Simonsen, L.; Viboud, C.; Miller, M.A.; Taylor, J.; George, K.S.; Griesemer, S.B.; Ghedin, E.; Sengamalay, N.A.; Spiro, D.J.; et al. Stochastic Processes Are Key Determinants of Short-Term Evolution in Influenza A Virus. PLoS Pathog. 2006, 2, e125. [Google Scholar] [CrossRef]
- Anderson, T.K.; Macken, C.A.; Lewis, N.S.; Scheuermann, R.H.; Van Reeth, K.; Brown, I.H.; Swenson, S.L.; Simon, G.; Saito, T.; Berhane, Y.; et al. A Phylogeny-Based Global Nomenclature System and Automated Annotation Tool for H1 Hemagglutinin Genes from Swine Influenza A Viruses. mSphere 2016, 1, e00275-16. [Google Scholar] [CrossRef] [Green Version]
- Scholtissek, C.; Bürger, H.; Bachmann, P.A.; Hannoun, C. Genetic relatedness of hemagglutinins of the H1 subtype of influenza a viruses isolated from swine and birds. Virology 1983, 129, 521–523. [Google Scholar] [CrossRef]
- De Jong, J.C.; Smith, D.J.; Lapedes, A.S.; Donatelli, I.; Campitelli, L.; Barigazzi, G.; VanReeth, K.; Jones, T.C.; Rimmelzwaan, G.F.; Osterhaus, A.D.M.E.; et al. Antigenic and genetic evolution of swine influenza A (H3N2) viruses in Europe. J. Virol. 2007, 81, 4315–4322. [Google Scholar] [CrossRef] [Green Version]
- Brown, I.H.; Chakraverty, P.; Harris, P.A.; Alexander, D.J. Disease outbreaks in pigs in Great Britain due to an influenza A virus of H1N2 subtype. Vet. Rec. 1995, 136, 328–329. [Google Scholar] [CrossRef]
- Pasma, T.; Joseph, T. Pandemic (H1N1) 2009 Infection in Swine Herds, Manitoba, Canada. Emerg. Infect. Dis. 2010, 16, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.; Larsen, L.E.; Dürrwald, R.; Foni, E.; Harder, T.; Van Reeth, K.; Markowska-Daniel, I.; Reid, S.M.; Dan, A.; Maldonado, J.; et al. European Surveillance Network for Influenza in Pigs: Surveillance Programs, Diagnostic Tools and Swine Influenza Virus Subtypes Identified in 14 European Countries from 2010 to 2013. PLoS ONE 2014, 9, e115815. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.J.; Langat, P.; Reid, S.M.; Lam, T.T.-Y.; Cotten, M.; Kelly, M.; Van Reeth, K.; Qiu, Y.; Simon, G.; Bonin, E.; et al. Molecular Epidemiology and Evolution of Influenza Viruses Circulating within European Swine between 2009 and 2013. J. Virol. 2015, 89, 9920–9931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trebbien, R.; Bragstad, K.; Larsen, L.E.; Nielsen, J.; Bøtner, A.; Heegaard, P.M.; Fomsgaard, A.; Viuff, B.; Hjulsager, C.K. Genetic and biological characterisation of an avian-like H1N2 swine influenza virus generated by reassortment of circulating avian-like H1N1 and H3N2 subtypes in Denmark. Virol. J. 2013, 10, 290. [Google Scholar] [CrossRef]
- Goecke, N.B.; Krog, J.S.; Hjulsager, C.K.; Skovgaard, K.; Harder, T.C.; Breum, S.Ø.; Larsen, L.E. Subtyping of Swine Influenza Viruses Using a High-Throughput Real-Time PCR Platform. Front. Cell. Infect. Microbiol. 2018, 8, 165. [Google Scholar] [CrossRef]
- Lange, J.; Groth, M.; Schlegel, M.; Krumbholz, A.; Wieczorek, K.; Ulrich, R.; Köppen, S.; Schulz, K.; Appl, D.; Selbitz, H.-J.; et al. Reassortants of the pandemic (H1N1) 2009 virus and establishment of a novel porcine H1N2 influenza virus, lineage in Germany. Vet. Microbiol. 2013, 167, 345–356. [Google Scholar] [CrossRef]
- Harder, T.C.; grosse Beilage, E.; Lange, E.; Meiners, C.; Döhring, S.; Pesch, S.; Noé, T.; Grund, C.; Beer, M.; Starick, E. Expanded Cocirculation of Stable Subtypes, Emerging Lineages, and New Sporadic Reassortants of Porcine Influenza Viruses in Swine Populations in Northwest Germany. J. Virol. 2013, 87, 10460–10476. [Google Scholar] [CrossRef] [Green Version]
- Pippig, J.; Ritzmann, M.; Büttner, M.; Neubauer-Juric, A. Influenza A Viruses Detected in Swine in Southern Germany after the H1N1 Pandemic in 2009. Zoonoses Public Health 2016, 63, 555–568. [Google Scholar] [CrossRef]
- Chiapponi, C.; Ebranati, E.; Pariani, E.; Faccini, S.; Luppi, A.; Baioni, L.; Manfredi, R.; Carta, V.; Merenda, M.; Affanni, P.; et al. Genetic analysis of human and swine influenza A viruses isolated in Northern Italy during 2010-2015. Zoonoses Public Health 2017, 65, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Zell, R.; Groth, M.; Krumbholz, A.; Lange, J.; Philipps, A.; Dürrwald, R. Displacement of the Gent/1999 human-like swine H1N2 influenza A virus lineage by novel H1N2 reassortants in Germany. Arch. Virol. 2020, 165, 55–67. [Google Scholar] [CrossRef]
- Henritzi, D.; Petric, P.P.; Lewis, N.S.; Graaf, A.; Pessia, A.; Starick, E.; Breithaupt, A.; Strebelow, G.; Luttermann, C.; Parker, L.M.K.; et al. Surveillance of European Domestic Pig Populations Identifies an Emerging Reservoir of Potentially Zoonotic Swine Influenza A Viruses. Cell Host Microbe 2020, 28, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Hervé, S.; Garin, E.; Calavas, D.; Lecarpentier, L.; Ngwa-Mbot, D.; Poliak, S.; Wendling, S.; Rose, N.; Simon, G. Virological and epidemiological patterns of swine influenza A virus infections in France: Cumulative data from the RESAVIP surveillance network, 2011–2018. Vet. Microbiol. 2019, 239, 108477. [Google Scholar] [CrossRef] [PubMed]
- Agreste. Les élevages de porcs en France métropolitaine en 2010. Primeur 2013, 300, 1–8. [Google Scholar]
- Marozin, S.; Gregory, V.; Cameron, K.; Bennett, M.; Valette, M.; Aymard, M.; Foni, E.; Barigazzi, G.; Lin, Y.; Hay, A. Antigenic and genetic diversity among swine influenza A H1N1 and H1N2 viruses in Europe. J. Gen. Virol. 2002, 83, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Chastagner, A.; Hervé, S.; Bonin, E.; Quéguiner, S.; Hirchaud, E.; Henritzi, D.; Béven, V.; Gorin, S.; Barbier, N.; Blanchard, Y.; et al. Spatio-temporal distribution and evolution of the A/H1N1 2009 pandemic virus in pigs in France from 2009 to 2017: Identification of a potential swine-specific lineage. J. Virol. 2018, 92, e00988-18. [Google Scholar] [CrossRef] [Green Version]
- Bonin, E.; Quéguiner, S.; Woudstra, C.; Gorin, S.; Barbier, N.; Harder, T.C.; Fach, P.; Hervé, S.; Simon, G. Molecular subtyping of European swine influenza viruses and scaling to high-throughput analysis. Virol. J. 2018, 15, 7. [Google Scholar] [CrossRef]
- Henritzi, D.; Zhao, N.; Starick, E.; Simon, G.; Krog, J.S.; Larsen, L.E.; Reid, S.M.; Brown, I.H.; Chiapponi, C.; Foni, E.; et al. Rapid detection and subtyping of European swine influenza viruses in porcine clinical samples by haemagglutinin- and neuraminidase-specific tetra- and triplex real-time RT-PCRs. Influenza Other Respir. Viruses 2016, 10, 504–517. [Google Scholar] [CrossRef] [Green Version]
- OIE. Chapter 3.8.7. Influenza A virus of swine. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2019; World Organization for Animal Health: Geneva, Switzerland, 2015; pp. 1–14. Available online: http://www.oie.int/en/international-standard-setting/terrestrial-manual/access-online/ (accessed on 12 November 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-Metagenomes from chimeric MDA products. J Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [Green Version]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome sequence assembly using trace signals and additional sequence information. Comput. Sci. Biol. Proc. Ger. Conf. Bioinforma. 1999, 99, 45–56. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, B.; Rambaut, A.; Drummond, A.J. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 2006, 23, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v 1.4.3. 2016. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 7 June 2017).
- Furuse, Y.; Suzuki, A.; Kamigaki, T.; Oshitani, H. Evolution of the M gene of the influenza A virus in different host species: Large-scale sequence analysis. Virol. J. 2009, 6, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Miotto, O.; Heiny, A.T.; Tan, T.W.; August, J.T.; Brusic, V. Identification of human-to-human transmissibility factors in PB2 proteins of influenza A by large-scale mutual information analysis. BMC Bioinform. 2008, 9, S18. [Google Scholar] [CrossRef] [Green Version]
- Sriwilaijaroen, N.; Suzuki, Y. Molecular basis of the structure and function of H1 hemagglutinin of influenza virus. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 226–249. [Google Scholar] [CrossRef] [Green Version]
- Lorusso, A.; Vincent, A.L.; Harland, M.L.; Alt, D.; Bayles, D.O.; Swenson, S.L.; Gramer, M.R.; Russell, C.A.; Smith, D.J.; Lager, K.M.; et al. Genetic and antigenic characterization of H1 influenza viruses from United States swine from 2008. J. Gen. Virol. 2011, 92, 919–930. [Google Scholar] [CrossRef]
- Colman, P.M.; Varghese, J.N.; Laver, W.G. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 1983, 303, 41–44. [Google Scholar] [CrossRef]
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza Virus Neuraminidase Structure and Functions. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chastagner, A.; Bonin, E.; Fablet, C.; Quéguiner, S.; Hirchaud, E.; Lucas, P.; Gorin, S.; Barbier, N.; Béven, V.; Garin, E.; et al. Virus persistence in pig herds led to successive reassortment events between swine and human influenza A viruses, resulting in the emergence of a novel triple-reassortant swine influenza virus. Vet. Res. 2019, 50, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Summary Table of Neuraminidase Amino Acid Substitutions Associated with Reduced Inhibition by Neuraminidase Inhibitors (NAI). Available online: https://www.who.int/influenza/gisrs_laboratory/antiviral_susceptibility/NAI_Reduced_Susceptibility_Marker_Table_WHO.pdf?ua=1 (accessed on 12 November 2020).
- Hervé, S.; Chastagner, A.; Quéguiner, S.; Barbier, N.; Gorin, S.; Blanchard, Y.; Jardin, A.; Dommergues, L.; Rose, N.; Simon, G. Identification of a novel swine influenza virus H1avN2 in several herds in Brittany. Bull. Épidémiologique, Santé Anim. Et Aliment. 2020, 89. in press. [Google Scholar]
- Pielak, R.M.; Schnell, J.R.; Chou, J.J. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. USA 2009, 106, 7379–7384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Ma, C.; Liu, X. PA-X: A key regulator of influenza A virus pathogenicity and host immune responses. Med. Microbiol. Immunol. 2018, 207, 255–269. [Google Scholar] [CrossRef]
- Kosik, I.; Holly, J.; Russ, G. PB1-F2 expedition from the whole protein through the domain to aa residue function. Acta Virol. 2013, 57, 138–148. [Google Scholar] [CrossRef]
- Seo, S.H.; Hoffmann, E.; Webster, R.G. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat. Med. 2002, 8, 950–954. [Google Scholar] [CrossRef]
- Plant, E.P.; Liu, T.M.; Xie, H.; Ye, Z. Mutations to A/Puerto Rico/8/34 PB1 gene improves seasonal reassortant influenza A virus growth kinetics. Vaccine 2012, 31, 207–212. [Google Scholar] [CrossRef]
- Xu, L.; Bao, L.; Zhou, J.; Wang, D.; Deng, W.; Lv, Q.; Ma, Y.; Li, F.; Sun, H.; Zhan, L.; et al. Genomic polymorphism of the pandemic A (H1N1) influenza viruses correlates with viral replication, virulence, and pathogenicity in vitro and in vivo. PLoS ONE 2011, 6, e20698. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Yamada, S.; Fukuyama, S.; Murakami, S.; Zhao, D.; Uraki, R.; Watanabe, T.; Tomita, Y.; Macken, C.; Neumann, G.; et al. Virulence-Affecting Amino Acid Changes in the PA Protein of H7N9 Influenza A Viruses. J. Virol. 2014, 88, 3127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, M.; Cooper, L.; Xu, X.; Thompson, W.; Krauss, S.; Guan, Y.; Zhou, N.; Klimov, A.; Cox, N.; Webster, R.; et al. Molecular changes associated with the transmission of avian influenza a H5N1 and H9N2 viruses to humans. J. Med. Virol. 2002, 66, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Manz, B.; Dornfeld, D.; Gotz, V.; Zell, R.; Zimmermann, P.; Haller, O.; Kochs, G.; Schwemmle, M. Pandemic influenza A viruses escape from restriction by human MxA through adaptive mutations in the nucleoprotein. PLoS Pathog. 2013, 9, e1003279. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Feng, Z.; Chen, Y.; Yang, L.; Liu, J.; Li, X.; Liu, S.; Zhou, L.; Wei, H.; Gao, R.; et al. Mammalian-adaptive mutation NP-Q357K in Eurasian H1N1 Swine Influenza viruses determines the virulence phenotype in mice. Emerg. Microbes Infect. 2019, 8, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonin, E.; Hervé, S.; Quéguiner, S.; Barbier, N.; Gorin, S.; Garin, E.; Wendling, S.; Simon, G. Distinction de plusieurs sous-populations de virus influenza porcins H1avN2 en France [Distinction of several subpopulations of H1avN2 swine influenza viruses in France]. Bull. Épidémiologique, Santé Anim. Et Aliment. 2016, 75, 11. [Google Scholar]
- Moreno, A.; Chiapponi, C.; Boniotti, M.B.; Sozzi, E.; Foni, E.; Barbieri, I.; Zanoni, M.G.; Faccini, S.; Lelli, D.; Cordioli, P. Genomic characterization of H1N2 swine influenza viruses in Italy. Vet. Microbiol. 2012, 156, 265–276. [Google Scholar] [CrossRef]
- Sobolev, I.; Kurskaya, O.; Leonov, S.; Kabilov, M.; Alikina, T.; Alekseev, A.; Yushkov, Y.; Saito, T.; Uchida, Y.; Mine, J.; et al. Novel reassortant of H1N1 swine influenza virus detected in pig population in Russia. Emerg. Microbes Infect. 2019, 8, 1456–1464. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.I.; Culhane, M.R.; Trovao, N.S.; Patnayak, D.P.; Halpin, R.A.; Lin, X.; Shilts, M.H.; Das, S.R.; Detmer, S.E. The emergence and evolution of influenza A (H1alpha) viruses in swine in Canada and the United States. J. Gen. Virol. 2017, 98, 2663–2675. [Google Scholar] [CrossRef]
- Deblanc, C.; Quéguiner, S.; Gorin, S.; Chastagner, A.; Hervé, S.; Paboeuf, F.; Simon, G. Evaluation of the Pathogenicity and the Escape from Vaccine Protection of a New Antigenic Variant Derived from the European Human-Like Reassortant Swine H1N2 Influenza Virus. Viruses 2020, 12, 1155. [Google Scholar] [CrossRef]
- Salines, M.; Andraud, M.; Rose, N. Pig movements in France: Designing network models fitting the transmission route of pathogens. PLoS ONE 2017, 12, e0185858. [Google Scholar] [CrossRef] [Green Version]
- Lewis, N.S.; Russell, C.A.; Langat, P.; Anderson, T.K.; Berger, K.; Bielejec, F.; Burke, D.F.; Dudas, G.; Fonville, J.M.; Fouchier, R.A.; et al. The global antigenic diversity of swine influenza A viruses. eLife 2016, 5, e12217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.J.; Lapedes, A.S.; de Jong, J.C.; Bestebroer, T.M.; Rimmelzwaan, G.F.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Mapping the Antigenic and Genetic Evolution of Influenza Virus. Science 2004, 305, 371–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedford, T.; Suchard, M.A.; Lemey, P.; Dudas, G.; Gregory, V.; Hay, A.J.; McCauley, J.W.; Russell, C.A.; Smith, D.J.; Rambaut, A. Integrating influenza antigenic dynamics with molecular evolution. eLife 2014, 3, e01914. [Google Scholar] [CrossRef] [PubMed]
- Abente, E.J.; Santos, J.; Lewis, N.S.; Gauger, P.C.; Stratton, J.; Skepner, E.; Anderson, T.K.; Rajao, D.S.; Perez, D.R.; Vincent, A.L. The Molecular Determinants of Antibody Recognition and Antigenic Drift in the H3 Hemagglutinin of Swine Influenza A Virus. J. Virol. 2016, 90, 8266–8280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaymard, A.; Le Briand, N.; Frobert, E.; Lina, B.; Escuret, V. Functional balance between neuraminidase and haemagglutinin in influenza viruses. Clin. Microbiol. Infect. 2016, 22, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.J.; Schepens, B.; Seok, J.H.; Kim, S.; Roose, K.; Lee, J.-H.; Gallardo, R.; Van Hamme, E.; Schymkowitz, J.; Rousseau, F.; et al. Structure of the Extracellular Domain of Matrix Protein 2 of Influenza A Virus in Complex with a Protective Monoclonal Antibody. J. Virol. 2015, 89, 3700–3711. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-J.; Lee, Y.-T.; Kim, M.-C.; Lee, Y.-N.; Kim, K.-H.; Ko, E.-J.; Song, J.-M.; Kang, S.-M. Cross-Protective Efficacy of Influenza Virus M2e Containing Virus-Like Particles Is Superior to Hemagglutinin Vaccines and Variable Depending on the Genetic Backgrounds of Mice. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Kim, K.-H.; Kwon, Y.-M.; Lee, Y.-T.; Kim, M.-C.; Hwang, H.; Ko, E.-J.; Lee, Y.; Choi, H.-J.; Kang, S.-M. Virus-Like Particles Are a Superior Platform for Presenting M2e Epitopes to Prime Humoral and Cellular Immunity against Influenza Virus. Vaccines 2018, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Makkoch, J.; Suwannakarn, K.; Payungporn, S.; Prachayangprecha, S.; Cheiocharnsin, T.; Linsuwanon, P.; Theamboonlers, A.; Poovorawan, Y. Whole genome characterization, phylogenetic and genome signature analysis of human pandemic H1N1 virus in Thailand, 2009–2012. PLoS ONE 2012, 7, e51275. [Google Scholar] [CrossRef]
- Weeks-Gorospe, J.N.; Hurtig, H.R.; Iverson, A.R.; Schuneman, M.J.; Webby, R.J.; McCullers, J.A.; Huber, V.C. Naturally Occurring Swine Influenza A Virus PB1-F2 Phenotypes That Contribute to Superinfection with Gram-Positive Respiratory Pathogens. J. Virol. 2012, 86, 9035–9043. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, M.; Basnet, S.; Burley, L.M.; Neumann, G.; Hatta, M.; Kawaoka, Y. Impact of amino acid mutations in PB2, PB1-F2, and NS1 on the replication and pathogenicity of pandemic (H1N1) 2009 influenza viruses. J. Virol. 2011, 85, 4596–4601. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain (Abbreviated Name) | Subtype | Swine H1 Clade [7] |
|---|---|---|
| A/Swine/Finistere/2899/82 (Fin2899/82) | H1avN1 | 1C.1 |
| A/Swine/Morbihan/0070/05 (Mo0070/05) | H1avN1 | 1C.2.1 |
| A/Swine/Cotes d’Armor/0388/09 (CA0388/09) | H1avN1 | 1C.2.1 |
| A/Swine/Cotes d’Armor/0186/10 (CA0186/10) | H1avN2 | 1C.2.1 |
| A/Swine/France/65-150242/15 (65-150242) | H1avN2 | 1C.2 |
| A/Swine/Scotland/410440/94 (Scot/94) | H1huN2 | 1B.1 |
| A/Swine/Cotes d’Armor/0214/06 (CA0214/06) | H1huN2 | 1B.1.1 |
| A/Swine/Cotes d’Armor/0113/06 (CA0113/06) | H1huN2 | 1B.1.2.3 |
| A/Swine/France/22-130212/13 (22-130212) | H1huN2 | 1B.1.2.3 (∆146-147) * |
| A/Swine/Cotes d’Armor/0070/10 (CA0070/10) | H1huN1 | 1B.1.2.3 |
| A/Swine/Cotes d’Armor/0190/06 (CA0190/06) | H1huN1 | 1B.1.2.3 (∆147) * |
| A/Swine/England/117316/86 | clswH1N1 | 1A.1-like |
| A/California/04/09 | H1N1pdm | 1A.3.3.2 |
| A/Swine/Sarthe/0255/10 | H1N1pdm | 1A.3.3.2 |
| Hemagglutination Inhibition Titer to Pig Antiserum Produced against Selected H1av Isolates | ||||||
|---|---|---|---|---|---|---|
| HA Clade | swIAV Strains (Name or Period of Isolation or Subgroup Within 1C.2.1 Clade of Tested Strains) | A/Sw/Finistere/2899/82 (1C.1-H1avN1) | A/Sw/Morbihan/0070/05 (1C.2.1-H1avN1) | A/Sw/Cotes d’Armor/0388/09 (1C.2.1-H1avN1) | A/Sw/Cotes d’Armor/0186/10 (1C.2.1-H1avN2) | A/Sw/France/65-150242/15 (1C.2-H1avN2) |
| 1C.1 | A/Sw/Finistere/2899/82 | 2560 | 80 | |||
| 1C.2.1 | A/Sw/Morbihan/0070/05 | 40 | 640 | |||
| A/Sw/Cotes d’Armor/0388/09 | 320 | 640 | ||||
| A/Sw/Cotes d’Armor/0186/10 | 320 | 320 | 320 | |||
| 1C.2 | A/Sw/France/65-150242/15 | 80 | <10 | 2560 | ||
| 1C.2.1 | 2000–2010 | 215.34 ± 2.85 a | 490.23 ± 1.62 a | 525.01 ± 1.69 a | ||
| [40–640] | [160–1280] | [320–1280] | ||||
| 16 | 26 | 7 | ||||
| 2011–2014 | 750.18 ± 1.58 b | 879.33 ± 1.83 b | 242.51 ± 1.81 c | |||
| [320–1280] | [320–2560] | [<10–640] | ||||
| 48 | 48 | 30 | ||||
| 2015–2018 | 336.99 ± 1.96 d | 132.81 ± 1.71 e | 16.59 ± 3.17 f | |||
| [40–1280] | [80–320] | [<10–80] | ||||
| 58 | 58 | 36 | ||||
| 1C.2 | 2015–2018 | 40 ± 3.32 | 320 ± 18.93 | |||
| [10–80] | [<10] | [40–2560] | ||||
| 3 | 3 | 2 | ||||
| 1C.2.2 | 2013–2015 | 160 ± 2.67 | 113.14 ± 1.63 | |||
| [80–320] | [80–160] | |||||
| 2 | 2 | |||||
| Subgroups within 1C.2.1 2015–2018 | group A | 427.15 ± 1.84 g | 146.72 ± 1.8 ij | 30.07 ± 1.74 k | ||
| [160–1280] | [80–320] | [10–80] | ||||
| 22 | 22 | 15 | ||||
| group B | 246.75 ± 1.43 h | 113.14 ± 1.45 i | 9.56 ± 3.66 m | |||
| [160–320] | [80–160] | [<10–20] | ||||
| 6 | 6 | 4 | ||||
| group C | 470.32 ± 1.44 g | 201.59 ± 1.63 j | 26.92 ± 1.45 k | |||
| [320–640] | [80–320] | [20–40] | ||||
| 8 | 8 | 6 | ||||
| Others (not A, B or C) | 265.52 ± 2.17 h | 110.16 ± 1.57 i | 7.7 ± 4.11 m | |||
| [40–1280] | [<10–320] | [<10–40] | ||||
| 22 | 22 | 11 | ||||
| HA Clade | swIAV Strains (Name or Period of Isolation or Subgroup within 1B.1.2.3 Clade of Tested Strains) | Hemagglutination Inhibition Titer to Pig Antiserum Produced Against Selected H1hu Isolates | |||||
|---|---|---|---|---|---|---|---|
| A/Sw/Scotland/ 410440/94 (1B.1-H1huN2) | A/Sw/Cotes d’Armor/0214/06 (1B.1.1-H1huN2) | A/Sw/Cotes d’Armor/0070/10 (1B.1.2.3-H1huN1) | A/Sw/Cotes d’Armor/0113/06 (1B.1.2.3-D H1huN2) | A/Sw/France/ 22-130212/13 (1B.1.2.3Δ146-147 H1huN2) | A/Sw/Cotes d’Armor/0190/06 (1B.1.2.3Δ147 H1huN1) | ||
| 1B.1 | A/Sw/Scotland/410440/94 | 2560 | 320 | 160 | 10 | ||
| 1B.1.1 | A/Sw/Cotes d’Armor/0214/06 | 320 | 1280 | 160 | 10 | ||
| 1B.1.2.3 | A/Sw/Cotes d’Armor/0070/10 | 1280 | 640 | ||||
| A/Sw/Cotes d’Armor/0113/06 | 1280 | 320 | 1280 | 20 | |||
| A/Sw/France/ 22-130212/13 | 160 | 320 | 320 | 40 | 1280 | 160 | |
| A/Sw/Cotes d’Armor/0190/06 | 40 | 10 | 20 | <10 | 1280 | ||
| 1B.1.1 | 2000–2010 | 320 ± 2.4 a [80–1280] 6 | 640 ± 1.63 a [320–1280] 5 | 100.79 ± 1.76 f [40–160] 6 | 4.57 ± 4.12 e [<10–20] 5 | ||
| 1B.1.2.3 | 2000–2010 | 1539.87 ± 1.63 b [640–2560] 15 | 359.19 ± 1.92 a,d [80–640] 12 | 452.55 ± 1.63 [320–640] 2 | 525.01 ± 1.88 d [160–1280] 14 | 40 ± 2.06 b [10–80] 12 | |
| 2011–2014 | 510.37 ± 3.87 a [40–5120] 49 | 187.44 ± 3.44 d [<10–1280] 38 | 240.8 ± 3.38 d [20–1280] 39 | 61.53 ± 6.27 f [<10–640] 50 | 320 ± 2 a,c [160–1280] 11 | 69.59 ± 3.88 f [<10–1280] 38 | |
| 2015–2018 | 499.65 ± 4.22 a,d [20–2560] 28 | 160 ± 1.76 [80–320] 4 | 342.97 ± 3.27 d [20–1280] 30 | 77.02 ± 4.81 f [<10–320] 30 | 201.59 ± 2.23 c [40–640] 30 | 134.54 ± 2.39 [40–320] 4 | |
| 1B.1.2.1 | 2015–2018 | 2031.87 ± 1.49 [1280–2560] 3 | 806.35 ± 1.49 [640–1280] 3 | 253.98 ± 1.49 [160–320] 3 | 126.99 ± 1.49 [80–160] 3 | ||
| Subgroups within 1B.1.2.3 2000–2010 | All except groups D | 1741.81 ± 1.65 g [640–2560] 9 | 390.08 ± 2.16 i [80–640] 7 | 452.55 ± 1.9 i [160–1280] 8 | 65.63 ± 1.4 p [40–80] 7 | ||
| group D | 1280 ± 1.55 g [640–2560] 6 | 320 ± 1.63 i [160–640] 5 | 640 ± 1.86 g,i [320–1280] 6 | 20 ± 1.63 n [10–40] 5 | |||
| Subgroups within 1B.1.2.3 2011–2014 | All except groups D & Δ146-147 | 1612.7 ± 1.66 g [640–5120] 21 | 452.55 ± 1.66 i [160–1280] 16 | 728.82 ± 1.68 j [320–1280] 16 | 181.49 ± 1.94 m [40–640] 22 | 190.27 ± 1.41 [160–320] 4 | 39.45 ± 3.07 p [<10-160] 16 |
| group D | 829.98 ± 2.84 g [80–2560] 8 | 12.65 ± 36.19 [<10–160] 2 | 160 ± 6.26 [20–640] 3 | 226.27 ± 2.67 i,m [40–640] 8 | 3.16 ± 5.09 [<10–10] 2 | ||
| Δ146-147 | 125.53 ± 1.76 h [40–320] 20 | 121.26 ± 1.99 h [40–320] 20 | 105.56 ± 2.21 h [20–320] 20 | 11.12 ± 5.17 k [<10–40] 20 | 430.69 ± 1.97 j [160–1280] 7 | 149.29 ± 2.1 h [40–1280] 20 | |
| Subgroups within 1B.1.2.32015–2018 | All except group Δ146-147 | 1280 ± 1.79 g [320–2560] 18 | 685.94 ± 1.81 j [160–1280] 20 | 190.27 ± 1.72 m [80–320] 20 | 144.2 ± 1.98 m [40–320] 20 | ||
| Δ146-147 | 91.9 ± 2.2 h [20–320] 10 | 126.99 ± 1.49 [80–160] 3 | 85.74 ± 2.14 h [20–160] 10 | 12.62 ± 3.92 k [<10–40] 10 | 393.97 ± 1.77 j [160–640] 10 | 201.59 ± 1.49 [160–320] 3 | |
| SwIAV Proteins | PB2 | PB1 | PB1-F2 | PA | PA-X | NP | M1 | M2 | NS1 | NS2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Protein length (in amino acid) | 759 | 757 | 90 | 716 | 260 | 498 | 252 | 97 | 230 | 121 |
| Nb of conserved residues (%) | 590 (77.7%) | 552 (72.9%) | 8 (8.9%) | 521 (72.8%) | 158 (60.8%) | 406 (81.5%) | 211 (83.7%) | 48 (49.5%) | 98 (42.6%) | 82 (67.8%) |
| Mean Entropy | 0.055 | 0.068 | 0.379 | 0.062 | 0.117 | 0.040 | 0.019 | 0.157 | 0.187 | 0.082 |
| Max. Entropy | 1.273 | 1.318 | 1.827 | 1.391 | 1.268 | 0.986 | 0.711 | 1.493 | 1.419 | 1.016 |
| Entropy st. dev. | 0.159 | 0.176 | 0.364 | 0.168 | 0.236 | 0.123 | 0.065 | 0.307 | 0.274 | 0.178 |
| Subtype | Protein-Encoding Genomic Segment | Frequency Nb (%) | Corresponding Genotype Previously Described in [13] * | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HA [7] | NA | PB2 | PB1 | PA | NP | M | NS | |||
| H1avN1 | 1C.2.1 | N1-EA | EA | EA | EA | EA | swEA | EA | 34 (21.1) | A |
| 1C.2.1 | N1-EA | EA | EA | EA | EA | swEU | EA | 55 (34.2) | A | |
| 1C.2.2 | N1-EA | EA | EA | EA | EA | swEA | EA | 3 (1.9) | A | |
| 1C.2.3 | N1-EA | EA | EA | EA | EA | swEU | EA | 2 (1.2) | A | |
| H1avN2 | 1C.2 | N2-Gent | EA | EA | EA | EA | Dk-like | EA | 2 (1.2) | D |
| 1C.2 | N2-Gent | pdm | pdm | pdm | pdm | pdm | pdm | 1 (0.6) | T | |
| 1C.2.1 | N2-Scot | EA | EA | EA | EA | swEA | EA | 3 (1.9) | G | |
| 1C.2.1 | N2-Scot | EA | EA | EA | EA | swEU | EA | 2 (1.2) | G | |
| 1C.2.1 | N2hu | EA | EA | EA | EA | swEA | EA | 1 (0.6) | I | |
| 1C.2.1 | N2hu | EA | EA | EA | EA | pdm | EA | 2 (1.2) | Not described | |
| H1huN2 | 1B.1.1 | N2-Scot | EA | EA | EA | EA | swEA | EA | 1 (0.6) | C |
| 1B.1.2.3 | N2-Scot | EA | EA | EA | EA | swEU | EA | 29 (18.0) | C | |
| 1B.1.2.3 | N2-Scot | EA | EA | EA | EA | swEA | EA | 9 (5.6) | C | |
| 1B.1.2.3 (Δ146-147) | N2-Scot | EA | EA | EA | EA | swEA | EA | 5 (3.1) | C | |
| 1B.1.2.3 (Δ146-147) | N2-Scot | EA | EA | EA | EA | swEU | EA | 4 (2.5) | C | |
| 1B.1.2.1 | N2-Gent | EA | EA | EA | EA | swEU | EA | 4 (2.5) | E | |
| H1huN1 | 1B.1.2.3 | N1-EA | EA | EA | EA | EA | swEU | EA | 4 (2.5) | H |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chastagner, A.; Hervé, S.; Quéguiner, S.; Hirchaud, E.; Lucas, P.; Gorin, S.; Béven, V.; Barbier, N.; Deblanc, C.; Blanchard, Y.; et al. Genetic and Antigenic Evolution of European Swine Influenza A Viruses of HA-1C (Avian-Like) and HA-1B (Human-Like) Lineages in France from 2000 to 2018. Viruses 2020, 12, 1304. https://doi.org/10.3390/v12111304
Chastagner A, Hervé S, Quéguiner S, Hirchaud E, Lucas P, Gorin S, Béven V, Barbier N, Deblanc C, Blanchard Y, et al. Genetic and Antigenic Evolution of European Swine Influenza A Viruses of HA-1C (Avian-Like) and HA-1B (Human-Like) Lineages in France from 2000 to 2018. Viruses. 2020; 12(11):1304. https://doi.org/10.3390/v12111304
Chicago/Turabian StyleChastagner, Amélie, Séverine Hervé, Stéphane Quéguiner, Edouard Hirchaud, Pierrick Lucas, Stéphane Gorin, Véronique Béven, Nicolas Barbier, Céline Deblanc, Yannick Blanchard, and et al. 2020. "Genetic and Antigenic Evolution of European Swine Influenza A Viruses of HA-1C (Avian-Like) and HA-1B (Human-Like) Lineages in France from 2000 to 2018" Viruses 12, no. 11: 1304. https://doi.org/10.3390/v12111304
APA StyleChastagner, A., Hervé, S., Quéguiner, S., Hirchaud, E., Lucas, P., Gorin, S., Béven, V., Barbier, N., Deblanc, C., Blanchard, Y., & Simon, G. (2020). Genetic and Antigenic Evolution of European Swine Influenza A Viruses of HA-1C (Avian-Like) and HA-1B (Human-Like) Lineages in France from 2000 to 2018. Viruses, 12(11), 1304. https://doi.org/10.3390/v12111304