Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2’-O-Methylation of Viral RNA



Abstract
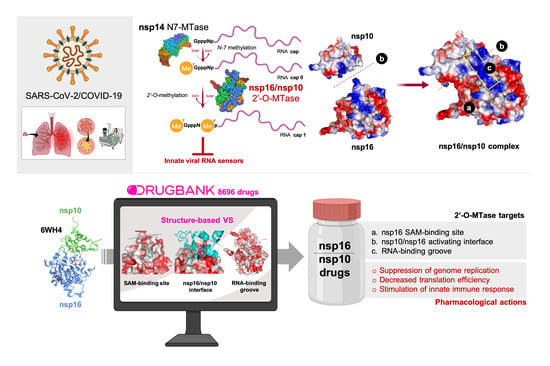
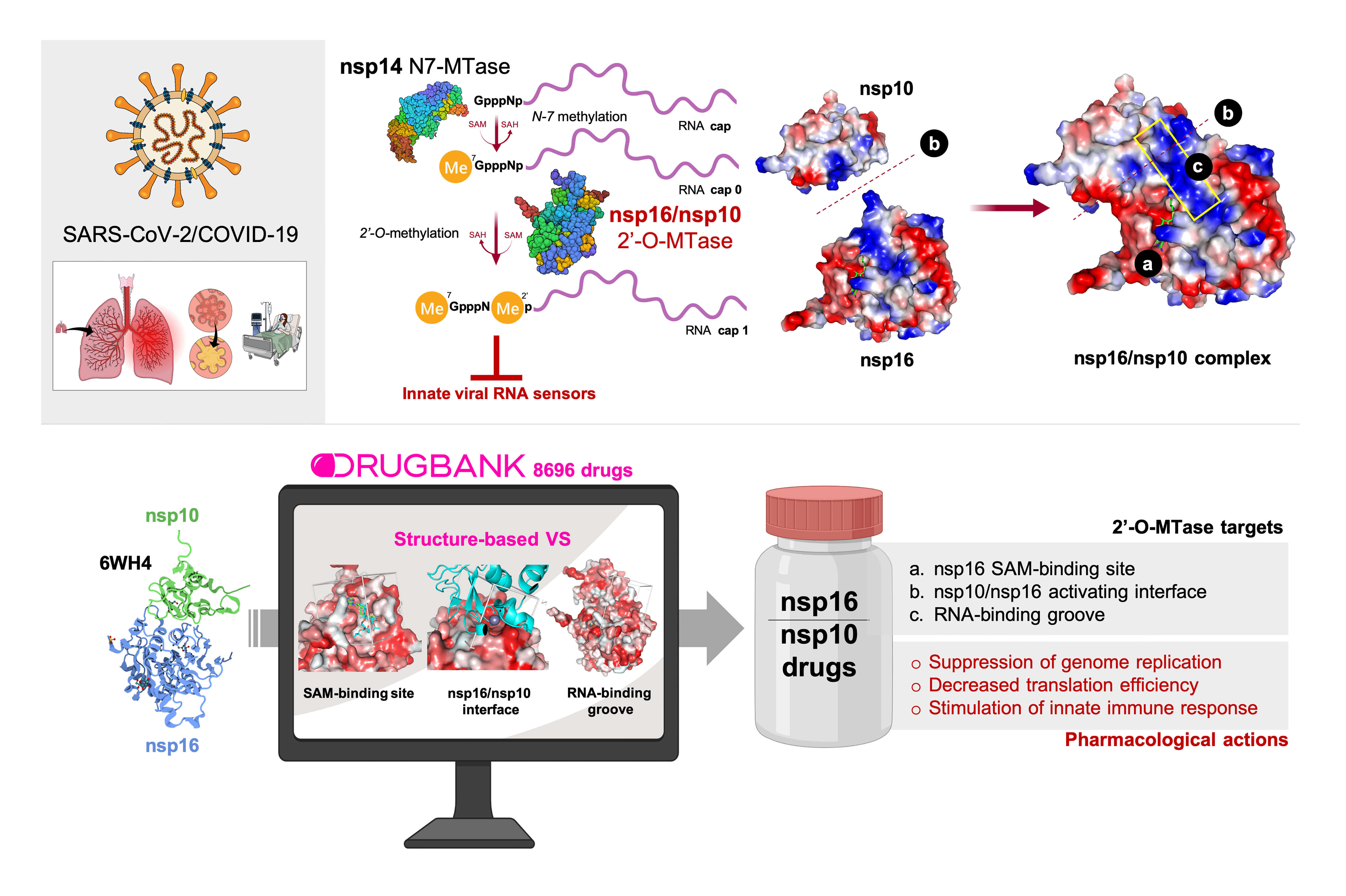
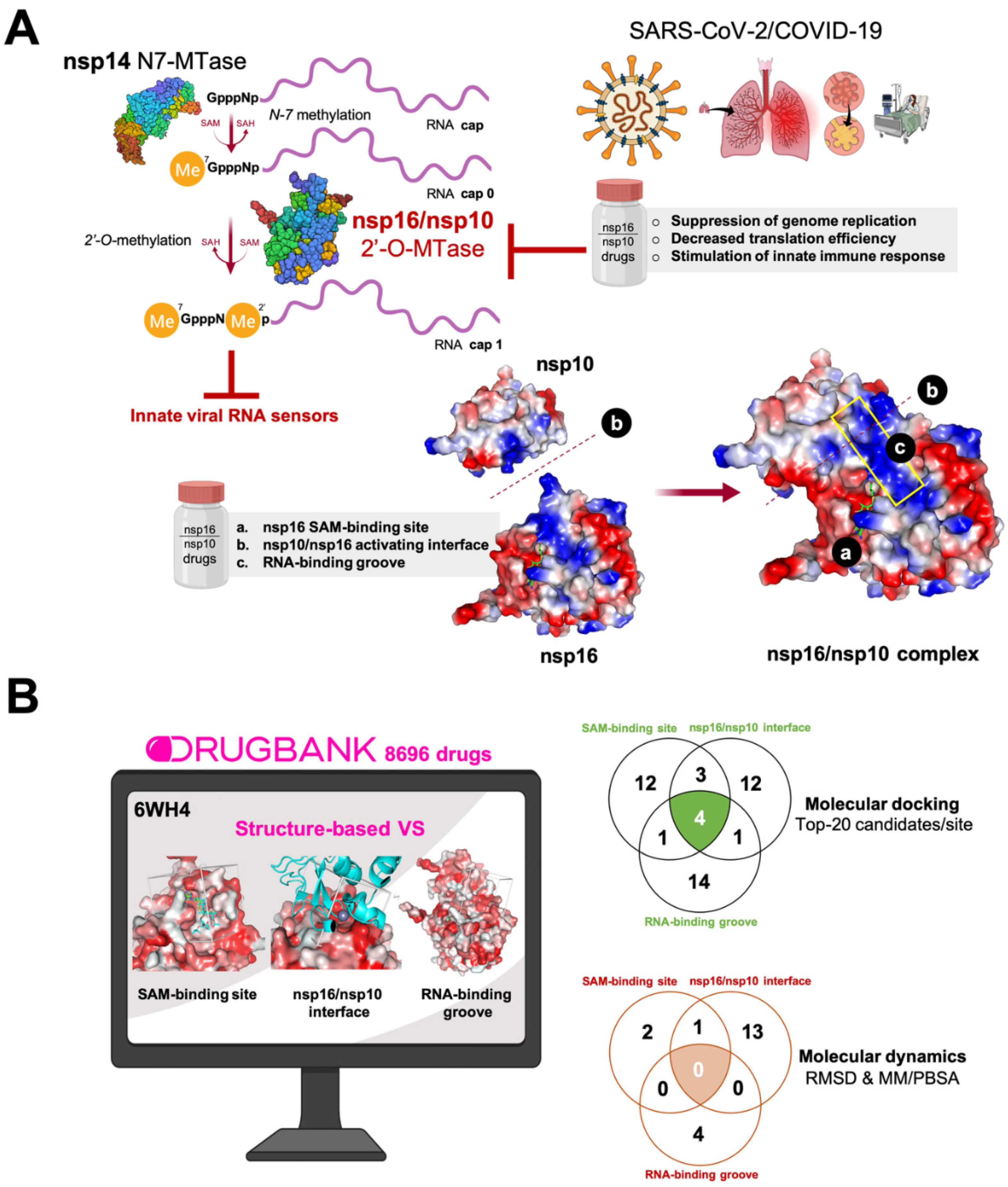
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drug Structures
2.2. Viral Proteins Structures
2.3. Molecular Docking Simulation
2.4. Molecular Dynamics Simulations
2.5. Evaluation of nsp16-Drug Interactions
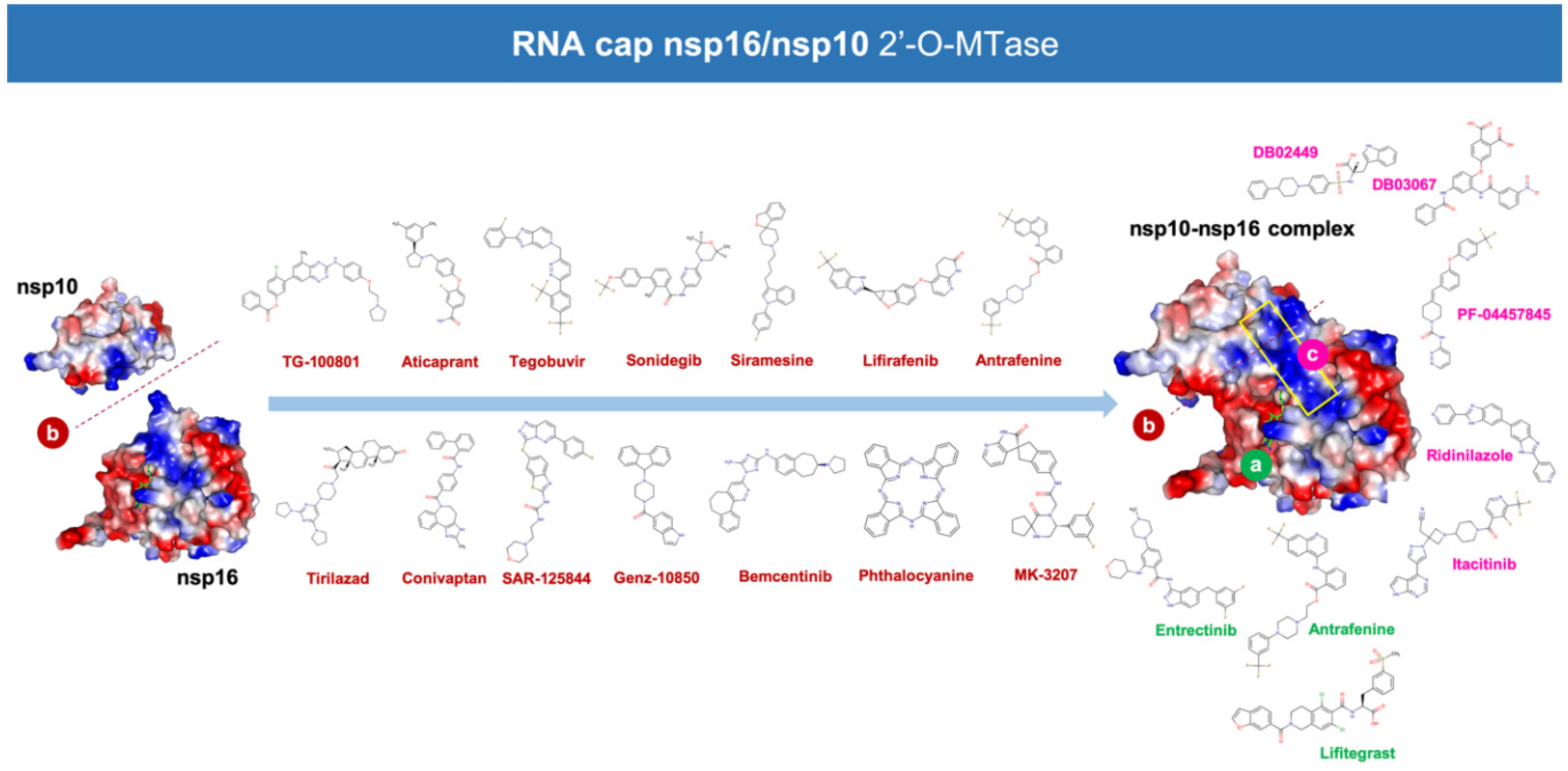
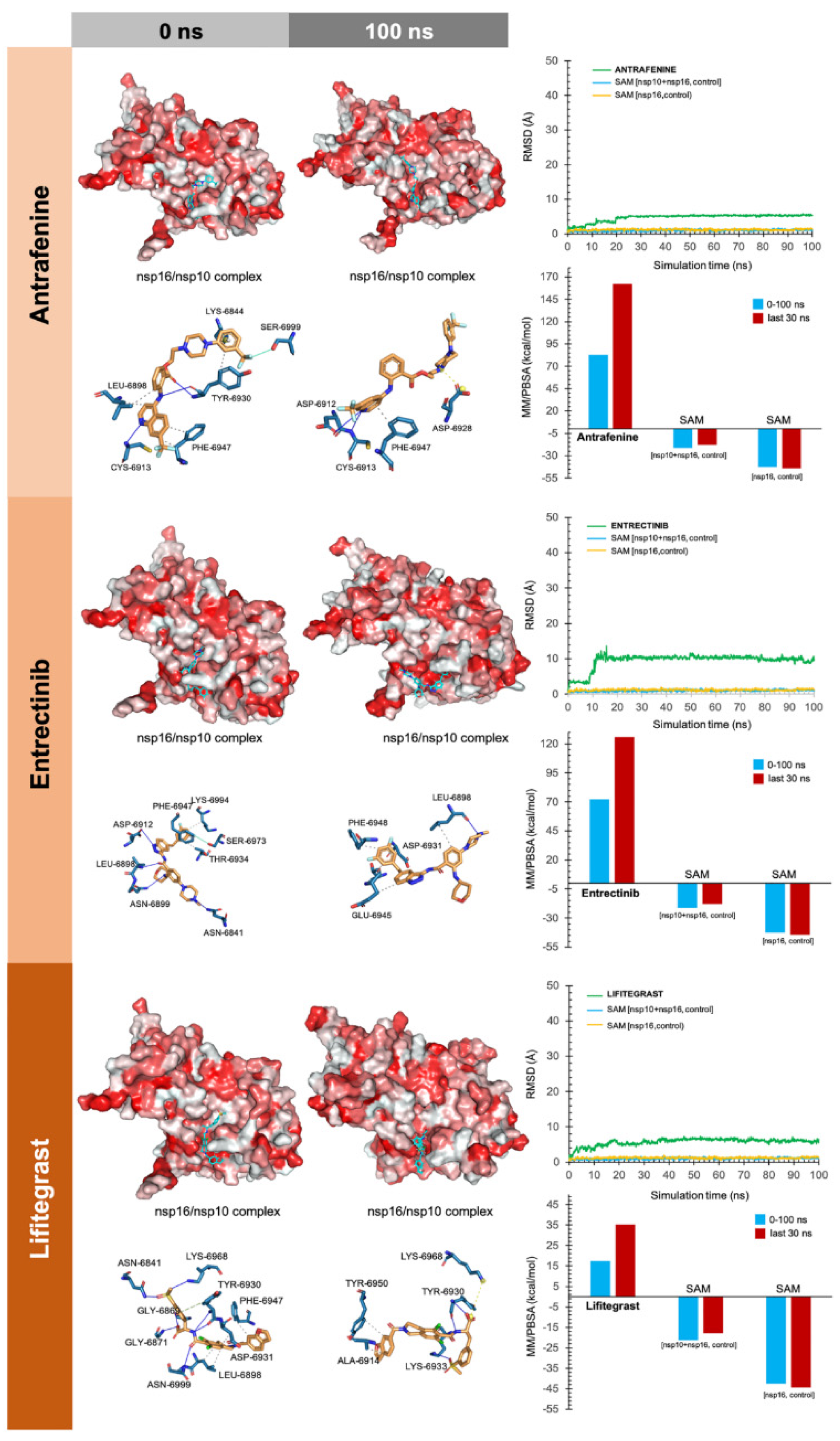
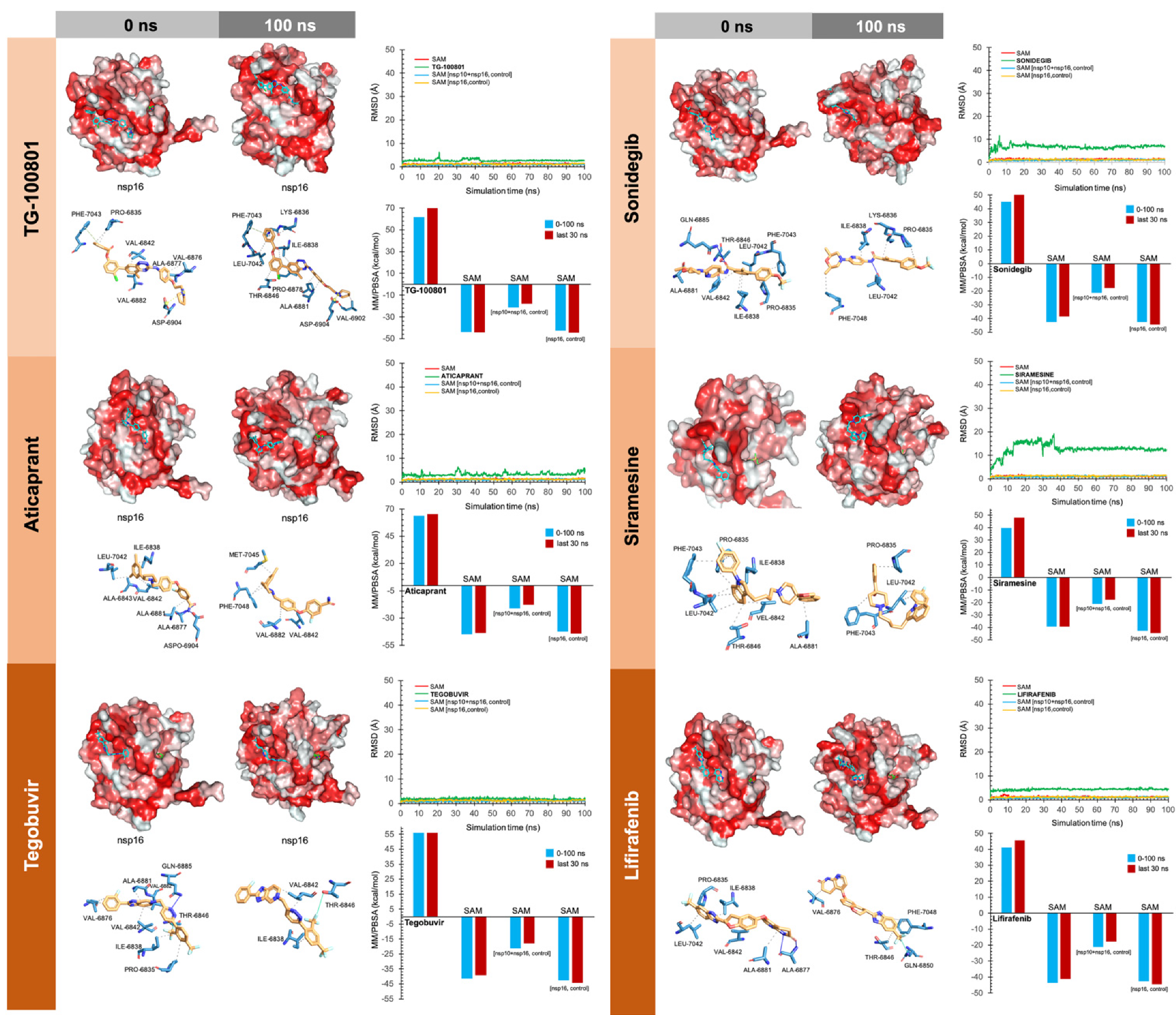
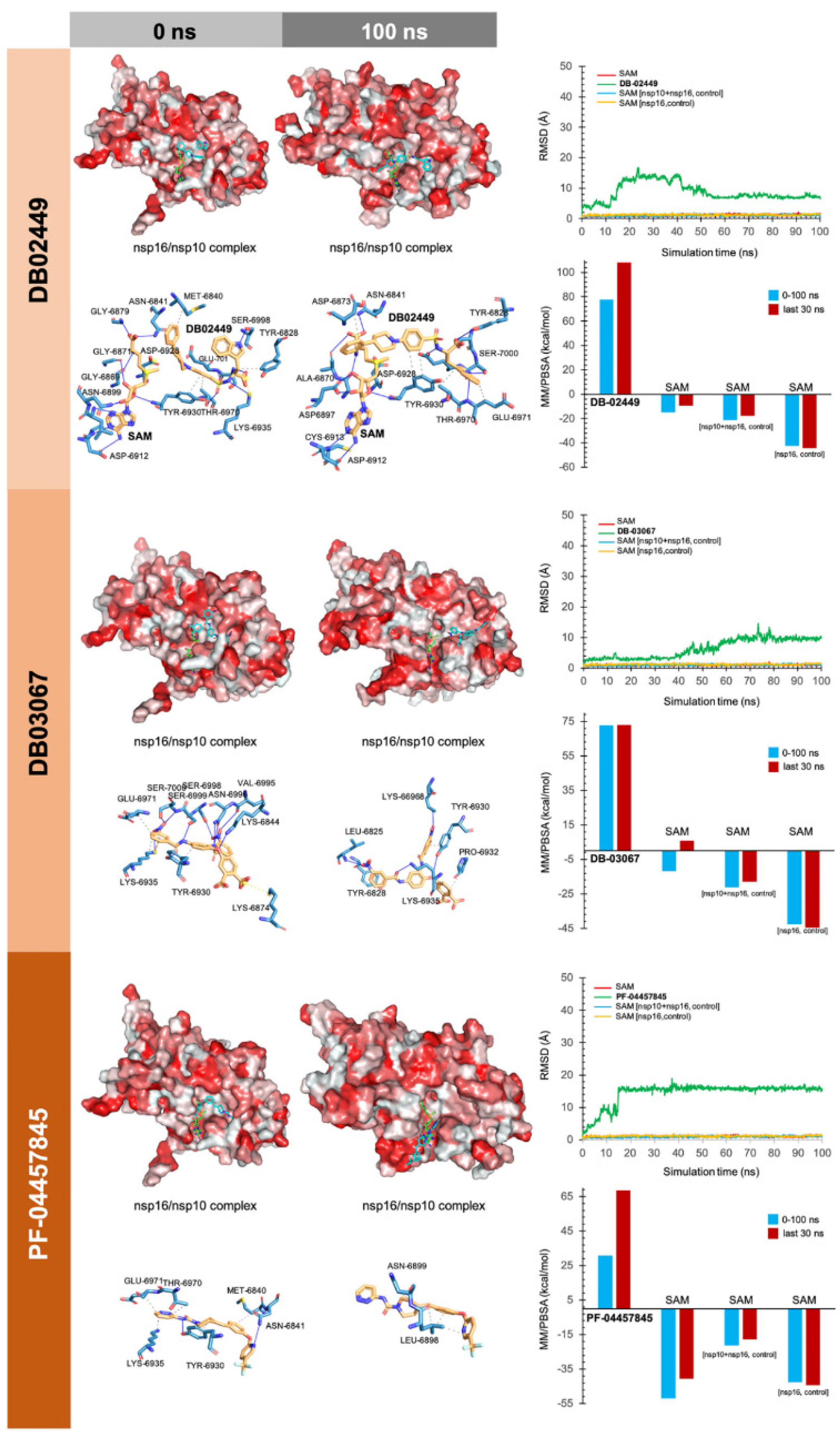
3. Results
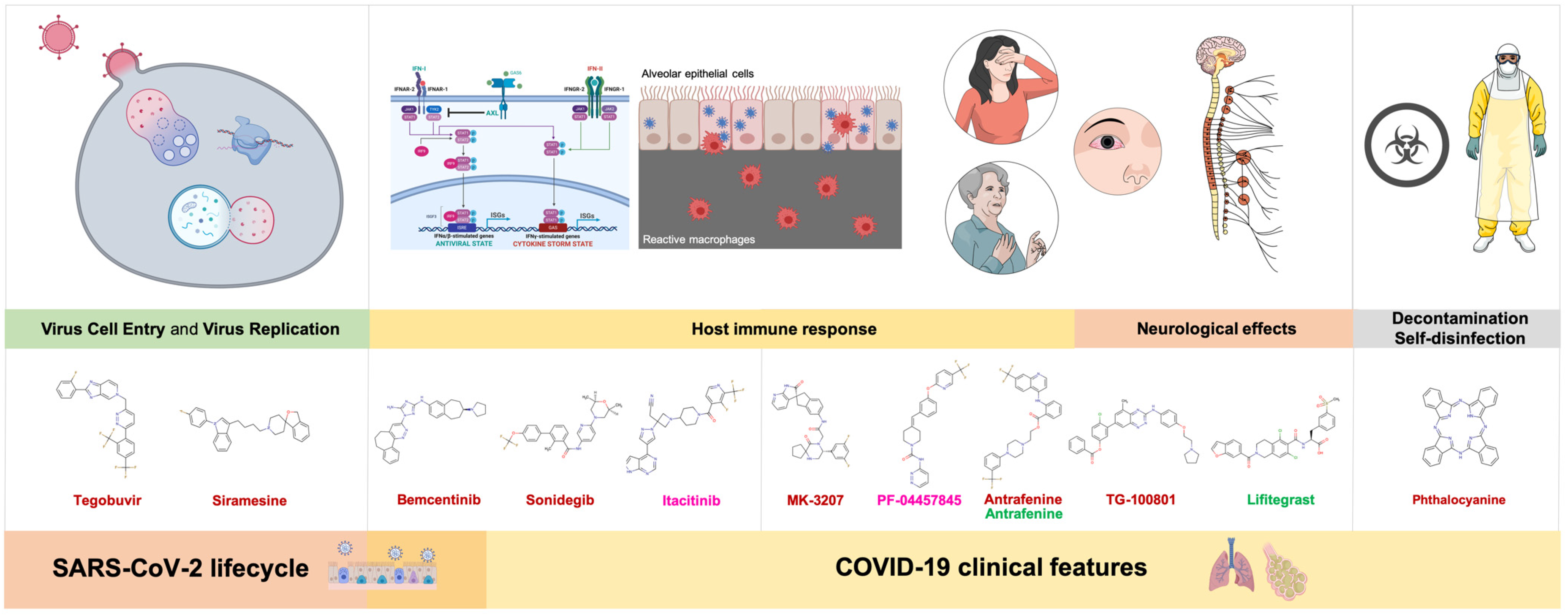
4. Discussion
4.1. Virus Cell Entry and Replication
4.1.1. Tegobuvir
4.1.2. Siramesine
4.2. Host Immune Response
4.2.1. Bemcentinib
4.2.2. Sonidegib
4.2.3. Itacitinib
4.3. Neurological Effects
4.3.1. MK-3207, PF-04457845, and Antrafenine
4.3.2. TG-100801 and Lifitegrast
4.4. Decontamination and Self-Disinfection
Phthalocyanine
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Wilder-Smith, A.; Chiew, C.J.; Lee, V.J. Can we contain the COVID-19 outbreak with the same measures as for SARS? Lancet Infect. Dis. 2020, 20, e102–e107. [Google Scholar] [CrossRef] [Green Version]
- Gates, B. Responding to Covid-19—A Once-in-a-Century Pandemic? N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Novel Coronavirus (2019-nCoV) Situation Reports. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 4 May 2020).
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schäfer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A. Compounds with therapeutic potential against novel respiratory 2019 coronavirus. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents. 2020, 105949. [Google Scholar] [CrossRef]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, ciaa237. [Google Scholar] [CrossRef] [Green Version]
- Touret, F.; de Lamballerie, X. Of chloroquine and COVID-19. Antivir. Res. 2020, 177, 104762. [Google Scholar] [CrossRef] [PubMed]
- Devaux, C.A.; Rolain, J.M.; Colson, P.; Raoult, D. New insights on the antiviral effects of chloroquine against coronavirus: What to expect for COVID-19? Int. J. Antimicrob. Agents. 2020, 105938. [Google Scholar] [CrossRef] [PubMed]
- Totura, A.L.; Bavari, S. Broad-spectrum coronavirus antiviral drug discovery. Expert Opin. Drug Discov. 2019, 14, 397–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017, 9, eaal3653. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. HLH Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Zhou, D.; Dai, S.M.; Tong, Q. COVID-19: A recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J. Antimicrob. Chemother. 2020, dkaa114. [Google Scholar] [CrossRef]
- Wu, D.; Yang, X.O. TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor Fedratinib. J. Microbiol. Immunol. Infect. 2020. [Google Scholar] [CrossRef]
- Boor, P.P.C.; de Ruiter, P.E.; Asmawidjaja, P.S.; Lubberts, E.; van der Laan, L.J.W.; Kwekkeboom, J. JAK-inhibitor tofacitinib suppresses interferon alfa production by plasmacytoid dendritic cells and inhibits arthrogenic and antiviral effects of interferon alfa. Transl. Res. 2017, 188, 67–79. [Google Scholar] [CrossRef]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93, e01815–e01818. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 71–280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R., Jr.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 2015, 116, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Matsuyama, S.; Li, X.; Takeda, M.; Kawaguchi, Y.; Inoue, J.I.; Matsuda, Z. Identification of Nafamostat as a Potent Inhibitor of Middle East Respiratory Syndrome Coronavirus S Protein-Mediated Membrane Fusion Using the Split-Protein-Based Cell-Cell Fusion Assay. Antimicrob. Agents Chemother. 2016, 60, 6532–6539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prussia, A.; Thepchatri, P.; Snyder, J.P.; Plemper, R.K. Systematic approaches towards the development of host-directed antiviral therapeutics. Int. J. Mol. Sci. 2011, 12, 4027–4052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Sisk, J.M.; Frieman, M.B.; Machamer, C.E. Coronavirus S protein-induced fusion is blocked prior to hemifusion by Abl kinase inhibitors. J. Gen. Virol. 2018, 99, 619–630. [Google Scholar] [CrossRef]
- Coleman, C.M.; Sisk, J.M.; Mingo, R.M.; Nelson, E.A.; White, J.M.; Frieman, M.B. Abelson Kinase Inhibitors Are Potent Inhibitors of Severe Acute Respiratory Syndrome Coronavirus and Middle East Respiratory Syndrome Coronavirus Fusion. J. Virol. 2016, 90, 8924–8933. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nukoolkarn, V.; Lee, V.S.; Malaisree, M.; Aruksakulwong, O.; Hannongbua, S. Molecular dynamic simulations analysis of ritonavir and lopinavir as SARS-CoV 3CL(pro) inhibitors. J. Theor. Biol. 2008, 254, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Stellbrink, H.J.; Arastéh, K.; Schürmann, D.; Stephan, C.; Dierynck, I.; Smyej, I.; Hoetelmans, R.M.; Truyers, C.; Meyvisch, P.; Jacquemyn, B.; et al. Antiviral activity, pharmacokinetics, and safety of the HIV-1 protease inhibitor TMC310911, coadministered with ritonavir, in treatment-naive HIV-1-infected patients. J. Acquir. Immune Defic. Syndr. 2014, 65, 283–289. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [Green Version]
- García-Sastre, A. 2 methylate or not 2 methylate: Viral evasion of the type I interferon response. Nat. Immunol. 2011, 12, 114–115. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010, 6, e1000863. [Google Scholar] [CrossRef]
- Lugari, A.; Betzi, S.; Decroly, E.; Bonnaud, E.; Hermant, A.; Guillemot, J.C.; Debarnot, C.; Borg, J.P.; Bouvet, M.; Canard, B.; et al. Molecular mapping of the RNA Cap 2′-O-methyltransferase activation interface between severe acute respiratory syndrome coronavirus nsp10 and nsp16. J. Biol. Chem. 2010, 285, 33230–33241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Su, C.; Ke, M.; Jin, X.; Xu, L.; Zhang, Z.; Wu, A.; Sun, Y.; Yang, Z.; Tien, P.; et al. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 2011, 7, e1002294. [Google Scholar] [CrossRef] [Green Version]
- Decroly, E.; Debarnot, C.; Ferron, F.; Bouvet, M.; Coutard, B.; Imbert, I.; Gluais, L.; Papageorgiou, N.; Sharff, A.; Bricogne, G.; et al. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2′-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog. 2011, 7, e1002059. [Google Scholar] [CrossRef] [Green Version]
- Ke, M.; Chen, Y.; Wu, A.; Sun, Y.; Su, C.; Wu, H.; Jin, X.; Tao, J.; Wang, Y.; Ma, X.; et al. Short peptides derived from the interaction domain of SARS coronavirus nonstructural protein nsp10 can suppress the 2′-O-methyltransferase activity of nsp10/nsp16 complex. Virus Res. 2012, 167, 322–328. [Google Scholar] [CrossRef]
- Jin, X.; Chen, Y.; Sun, Y.; Zeng, C.; Wang, Y.; Tao, J.; Wu, A.; Yu, X.; Zhang, Z.; Tian, J.; et al. Characterization of the guanine-N7 methyltransferase activity of coronavirus nsp14 on nucleotide GTP. Virus Res. 2013, 176, 45–52. [Google Scholar] [CrossRef]
- Chen, Y.; Tao, J.; Sun, Y.; Wu, A.; Su, C.; Gao, G.; Cai, H.; Qiu, S.; Wu, Y.; Ahola, T.; et al. Structure-function analysis of severe acute respiratory syndrome coronavirus RNA cap guanine-N7-methyltransferase. J. Virol. 2013, 87, 6296–6305. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, Z.; Tao, J.; Wang, Y.; Wu, A.; Yang, Z.; Wang, K.; Shi, L.; Chen, Y.; Guo, D. Yeast-based assays for the high-throughput screening of inhibitors of coronavirus RNA cap guanine-N7-methyltransferase. Antivir. Res. 2014, 104, 156–164. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L., Jr.; Josset, L.; Gralinski, L.E.; Scobey, T.; Agnihothram, S.; Katze, M.G.; Baric, R.S. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-o-methyltransferase activity. J. Virol. 2014, 88, 4251–4264. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Torres, V.; Losada-Echeberría, M.; Herranz-López, M.; Barrajón-Catalán, E.; Galiano, V.; Micol, V.; Encinar, J.A. New Mammalian Target of Rapamycin (mTOR) Modulators Derived from Natural Product Databases and Marine Extracts by Using Molecular Docking Techniques. Mar. Drugs. 2018, 16, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Cuyàs, E.; Verdura, S.; Lozano-Sánchez, J.; Viciano, I.; Llorach-Parés, L.; Nonell-Canals, A.; Bosch-Barrera, J.; Brunet, J.; Segura-Carretero, A.; Sanchez-Martinez, M.; et al. The extra virgin olive oil phenolic oleacein is a dual substrate-inhibitor of catechol-O-methyltransferase. Food Chem. Toxicol. 2019, 128, 35–45. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging 2020, 12, 8–34. [Google Scholar] [CrossRef] [Green Version]
- Cuyàs, E.; Gumuzio, J.; Verdura, S.; Brunet, J.; Bosch-Barrera, J.; Martin-Castillo, B.; Alarcón, T.; Encinar, J.A.; Martin, Á.G.; Menendez, J.A. The LSD1 inhibitor iadademstat (ORY-1001) targets SOX2-driven breast cancer stem cells: A potential epigenetic therapy in luminal-B and HER2-positive breast cancer subtypes. Aging 2020, 12, 4794. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Nguyen, P.H.; Pham, K.; Huynh, D.; Le, T.B.; Wang, H.; Ren, P.; Luo, R. Calculating protein-ligand binding affinities with MMPBSA: Method and error analysis. J. Comput. Chem. 2016, 37, 2436–2446. [Google Scholar] [CrossRef] [Green Version]
- Hyde, J.L.; Diamond, M.S. Innate immune restriction and antagonism of viral RNA lacking 2′-O methylation. Virology 2015, 479, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, S.; Cui, H.; Gao, Z.; Liu, M.; Lu, S.; Mkandawire, W.; Narykov, O.; Sun, M.; Korkin, D. Structural Genomics of SARS-CoV-2 Indicates Evolutionary Conserved Functional Regions of Viral Proteins. Viruses 2020, 12, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hebner, C.M.; Han, B.; Brendza, K.M.; Nash, M.; Sulfab, M.; Tian, Y.; Hung, M.; Fung, W.; Vivian, R.W.; Trenkle, J.; et al. The HCV non-nucleoside inhibitor Tegobuvir utilizes a novel mechanism of action to inhibit NS5B polymerase function. PLoS ONE 2012, 7, e39163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyles, D.L.; Rodriguez-Torres, M.; Lawitz, E.; Shiffman, M.L.; Pol, S.; Herring, R.W.; Massetto, B.; Kanwar, B.; Trenkle, J.D.; Pang, P.S.; et al. All-oral combination of ledipasvir, vedroprevir, tegobuvir, and ribavirin in treatment-naïve patients with genotype 1 HCV infection. Hepatology 2014, 60, 56–64. [Google Scholar] [CrossRef]
- Vliegen, I.; Paeshuyse, J.; Zhong, W.; Neyts, J. In vitro combinations containing Tegobuvir are highly efficient in curing cells from HCV replicon and in delaying/preventing the development of drug resistance. Antivir. Res. 2015, 120, 112–121. [Google Scholar] [CrossRef]
- Liu, M.; Xu, Q.; Guo, S.; Zuo, R.; Hong, Y.; Luo, Y.; Li, Y.; Gong, P.; Liu, Y. Design, synthesis, and structure-activity relationships of novel imidazo [4,5-c] pyridine derivatives as potent non-nucleoside inhibitors of hepatitis C virus NS5B. Bioorg Med Chem. 2018, 26, 2621–2631. [Google Scholar] [CrossRef]
- Ruan, Z.; Liu, C.; Guo, Y.; He, Z.; Huang, X.; Jia, X.; Yang, T. Potential Inhibitors Targeting RNA-Dependent RNA Polymerase Activity (NSP12) of SARS-CoV-2. Preprints 2020, 2020030024. [Google Scholar] [CrossRef]
- Khalili, J.S.; Zhu, H.; Mak, A.; Yan, Y.; Zhu, Y. Novel coronavirus treatment with ribavirin: Groundwork for evaluation concerning COVID-19. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020, 117592. [Google Scholar] [CrossRef] [PubMed]
- Ferron, F.; Subissi, L.; Silveira De Morais, A.T.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. USA 2018, 115, E162–E171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostenfeld, M.S.; Fehrenbacher, N.; Høyer-Hansen, M.; Thomsen, C.; Farkas, T.; Jäättelä, M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005, 65, 8975–8983. [Google Scholar] [CrossRef] [Green Version]
- Ostenfeld, M.S.; Høyer-Hansen, M.; Bastholm, L.; Fehrenbacher, N.; Olsen, O.D.; Groth-Pedersen, L.; Puustinen, P.; Kirkegaard-Sørensen, T.; Nylandsted, J.; Farkas, T.; et al. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy 2008, 4, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Zhitomirsky, B.; Yunaev, A.; Kreiserman, R.; Kaplan, A.; Stark, M.; Assaraf, Y.G. Lysosomotropic drugs activate TFEB via lysosomal membrane fluidization and consequent inhibition of mTORC1 activity. Cell Death Dis. 2018, 9, 1191. [Google Scholar] [CrossRef]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [Green Version]
- Villalpando-Rodriguez, G.E.; Blankstein, A.R.; Konzelman, C.; Gibson, S.B. Lysosomal Destabilizing Drug Siramesine and the Dual Tyrosine Kinase Inhibitor Lapatinib Induce a Synergistic Ferroptosis through Reduced Heme Oxygenase-1 (HO-1) Levels. Oxid. Med. Cell. Longev. 2019, 2019, 9561281. [Google Scholar] [CrossRef]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019, 10, 5770. [Google Scholar] [CrossRef]
- Hirata, Y.; Yamamoto, H.; Atta, M.S.; Mahmoud, S.; Oh-hashi, K.; Kiuchi, K. Chloroquine inhibits glutamate-induced death of a neuronal cell line by reducing reactive oxygen species through sigma-1 receptor. J. Neurochem. 2011, 119, 839–847. [Google Scholar] [CrossRef]
- Tesei, A.; Cortesi, M.; Zamagni, A.; Arienti, C.; Pignatta, S.; Zanoni, M.; Paolillo, M.; Curti, D.; Rui, M.; Rossi, D.; et al. Sigma Receptors as Endoplasmic Reticulum Stress “Gatekeepers” and their Modulators as Emerging New Weapons in the Fight against Cancer. Front. Pharmacol. 2018, 9, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Shen, H.M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liu, D.X. Coronavirus infection, ER stress, apoptosis and innate immunity. Front. Microbiol. 2014, 5, 296. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liao, Y.; Liu, D.X. Regulation of Stress Responses and Translational Control by Coronavirus. Viruses 2016, 8, 184. [Google Scholar] [CrossRef]
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- Heading, C. Siramesine H Lundbeck. Curr. Opin. Investig. Drugs. 2001, 2, 266–270. [Google Scholar]
- Søby, K.K.; Mikkelsen, J.D.; Meier, E.; Thomsen, C. Lu 28-179 labels a sigma(2)-site in rat and human brain. Neuropharmacology 2002, 43, 95–100. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Betz, R.M.; Dror, R.O.; Kruse, A.C. Structural basis for σ1 receptor ligand recognition. Nat. Struct. Mol. Biol. 2018, 25, 981–987. [Google Scholar] [CrossRef]
- Sahn, J.J.; Mejia, G.L.; Ray, P.R.; Martin, S.F.; Price, T.J. Sigma 2 Receptor/Tmem97 Agonists Produce Long Lasting Antineuropathic Pain Effects in Mice. ACS Chem. Neurosci. 2017, 8, 1801–1811. [Google Scholar] [CrossRef] [Green Version]
- Georgiadis, M.O.; Karoutzou, O.; Foscolos, A.S.; Papanastasiou, I. Sigma Receptor (σR) Ligands with Antiproliferative and Anticancer Activity. Molecules 2017, 22, 1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadahiro, H.; Kang, K.D.; Gibson, J.T.; Minata, M.; Yu, H.; Shi, J.; Chhipa, R.; Chen, Z.; Lu, S.; Simoni, Y.; et al. Activation of the Receptor Tyrosine Kinase AXL Regulates the Immune Microenvironment in Glioblastoma. Cancer Res. 2018, 78, 3002–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, K.F.; Du, W.; Sorrelle, N.B.; Wnuk-Lipinska, K.; Topalovski, M.; Toombs, J.E.; Cruz, V.H.; Yabuuchi, S.; Rajeshkumar, N.V.; Maitra, A.; et al. Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res. 2018, 78, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landolt, L.; Furriol, J.; Babickova, J.; Ahmed, L.; Eikrem, Ø.; Skogstrand, T.; Scherer, A.; Suliman, S.; Leh, S.; Lorens, J.B.; et al. AXL targeting reduces fibrosis development in experimental unilateral ureteral obstruction. Physiol. Rep. 2019, 7, e14091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutusaus, A.; de Gregorio, E.; Cucarull, B.; Cristóbal, H.; Aresté, C.; Graupera, I.; Coll, M.; Colell, A.; Gausdal, G.; Lorens, J.B.; et al. A Functional Role of GAS6/TAM in Nonalcoholic Steatohepatitis Progression Implicates AXL as Therapeutic Target. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 349–368. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef]
- Persaud, M.; Martinez-Lopez, A.; Buffone, C.; Porcelli, S.A.; Diaz-Griffero, F. Infection by Zika viruses requires the transmembrane protein AXL, endocytosis and low pH. Virology 2018, 518, 301–312. [Google Scholar] [CrossRef]
- Chen, F.; Song, Q.; Yu, Q. Axl inhibitor R428 induces apoptosis of cancer cells by blocking lysosomal acidification and recycling independent of Axl inhibition. Am. J. Cancer Res. 2018, 8, 1466–1482. [Google Scholar]
- Lotsberg, M.L.; Wnuk-Lipinska, K.; Terry, S.; Tan, T.Z.; Lu, N.; Trachsel-Moncho, L.; Røsland, G.V.; Siraji, M.I.; Hellesøy, M.; Rayford, A.; et al. AXL Targeting Abrogates Autophagic Flux and Induces Immunogenic Cell Death in Drug-Resistant Cancer Cells. J. Thorac. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burness, C.B. Sonidegib: First Global Approval. Drugs 2015, 75, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Collier, N.J.; Ali, F.R.; Lear, J.T. The safety and efficacy of sonidegib for the treatment of locally advanced basal cell carcinoma. Expert Rev. Anticancer Ther. 2016, 16, 1011–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Song, R.; Xie, J. Sonidegib: Mechanism of action, pharmacology, and clinical utility for advanced basal cell carcinomas. Onco Targets Ther. 2017, 10, 1645–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smelkinson, M.G. The Hedgehog Signaling Pathway Emerges as a Pathogenic Target. J. Dev. Biol. 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef]
- Gong, X.; Darpo, B.; Xue, H.; Punwani, N.; He, K.; Barbour, A.M.; Epstein, N.; Landman, R.; Chen, X.; Yeleswaram, S. Evaluation of Clinical Cardiac Safety of Itacitinib, a JAK1 Inhibitor, in Healthy Participants. Clin. Pharmacol. Drug Dev. 2019. [Google Scholar] [CrossRef]
- Bissonnette, R.; Luchi, M.; Fidelus-Gort, R.; Jackson, S.; Zhang, H.; Flores, R.; Newton, R.; Scherle, P.; Yeleswaram, S.; Chen, X.; et al. A randomized, double-blind, placebo-controlled, dose-escalation study of the safety and efficacy of INCB039110, an oral janus kinase 1 inhibitor, in patients with stable, chronic plaque psoriasis. J. Dermatolog. Treat. 2016, 27, 332–338. [Google Scholar] [CrossRef]
- Kettle, J.G.; Åstrand, A.; Catley, M.; Grimster, N.P.; Nilsson, M.; Su, Q.; Woessner, R. Inhibitors of JAK-family kinases: An update on the patent literature 2013–2015, part 1. Expert Opin. Ther. Pat. 2017, 27, 127–143. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef]
- Virtanen, A.T.; Haikarainen, T.; Raivola, J.; Silvennoinen, O. Selective JAKinibs: Prospects in Inflammatory and Autoimmune Diseases. BioDrugs 2019, 33, 15–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadina, M.; Le, M.T.; Schwartz, D.M.; Silvennoinen, O.; Nakayamada, S.; Yamaoka, K.; O’Shea, J.J. Janus kinases to jakinibs: From basic insights to clinical practice. Rheumatology 2019, 58, i4–i16. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Frigault, M.J.; Maziarz, R.T.; Naim, A.; Burke, L.; Tian, C.; Porter, D.L. Trial in Progress: A Phase 2, Single-Arm, Open-Label Study of Itacitinib (ITA) for the Prevention of Chimeric Antigen Receptor (CAR) T-Cell–Induced Cytokine Release Syndrome (CRS). Biol. Blood Marrow Transplant. 2020, 16, S269. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer. 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunologic features in severe and moderate Coronavirus Disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, S.F.; Ho, Y.C. SARS-CoV-2: A Storm is Raging. J. Clin Investig. 2020, 130, 137647. [Google Scholar] [CrossRef]
- Favalli, E.G.; Ingegnoli, F.; De Lucia, O.; Cincinelli, G.; Cimaz, R.; Caporali, R. COVID-19 infection and rheumatoid arthritis: Faraway, so close! Autoimmun. Rev. 2020, 19, 102523. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef] [Green Version]
- Bell, I.M.; Gallicchio, S.N.; Wood, M.R.; Quigley, A.G.; Stump, C.A.; Zartman, C.B.; Fay, J.F.; Li, C.C.; Lynch, J.J.; Moore, E.L.; et al. Discovery of MK-3207: A Highly Potent, Orally Bioavailable CGRP Receptor Antagonist. ACS Med. Chem. Lett. 2010, 1, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, D.J.; Aurora, S.K.; Dodick, D.W.; Goadsby, P.J.; Ge, Y.J.; Bachman, R.; Taraborelli, D.; Fan, X.; Assaid, C.; Lines, C.; et al. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia 2011, 31, 712–722. [Google Scholar] [CrossRef]
- Crowley, B.M.; Stump, C.A.; Nguyen, D.N.; Potteiger, C.M.; McWherter, M.A.; Paone, D.V.; Quigley, A.G.; Bruno, J.G.; Cui, D.; Culberson, J.C.; et al. Novel oxazolidinone calcitonin gene-related peptide (CGRP) receptor antagonists for the acute treatment of migraine. Bioorg. Med. Chem. Lett. 2015, 25, 4777–4781. [Google Scholar] [CrossRef] [PubMed]
- Negro, A.; Martelletti, P. Gepants for the treatment of migraine. Expert Opin. Investig. Drugs. 2019, 28, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Tepper, S.J. History and Review of anti-Calcitonin Gene-Related Peptide (CGRP) Therapies: From Translational Research to Treatment. Headache 2018, 58 (Suppl. 3), 238–275. [Google Scholar] [CrossRef] [PubMed]
- De Vries, T.; Villalón, C.M.; MaassenVanDenBrink, A. Pharmacological treatment of migraine: CGRP and 5-HT beyond the triptans. Pharmacol. Ther. 2020, 107528. [Google Scholar] [CrossRef]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host-Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020. [Google Scholar] [CrossRef]
- Han, H.; Xie, L.; Liu, R.; Yang, J.; Liu, F.; Wu, K.; Chen, L.; Hou, W.; Feng, Y.; Zhu, C. Analysis of heart injury laboratory parameters in 273 COVID-19 patients in one hospital in Wuhan, China. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Fried, J.A.; Ramasubbu, K.; Bhatt, R.; Topkara, V.K.; Clerkin, K.J.; Horn, E.; Rabbani, L.; Brodie, D.; Jain, S.S.; Kirtane, A.; et al. The Variety of Cardiovascular Presentations of COVID-19. Circulation 2020. [Google Scholar] [CrossRef] [Green Version]
- Bansal, M. Cardiovascular disease and COVID-19. Diabetes Metab. Syndr. 2020, 14, 247–250. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities in the novel Wuhan coronavirus (COVID-19) infection: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; He, L.; Zhang, Q.; Huang, Z.; Che, X.; Hou, J.; Wang, H.; Shen, H.; Qiu, L.; Li, Z.; et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: Implications for pathogenesis and virus transmission pathways. J. Pathol. 2004, 203, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Netland, J.; Meyerholz, D.K.; Moore, S.; Cassell, M.; Perlman, S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 2008, 82, 7264–7275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Mével, J.C.; Lancien, F.; Mimassi, N.; Conlon, J.M. Brain neuropeptides in central ventilatory and cardiovascular regulation in trout. Front. Endocrinol. 2012, 3, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dakhama, A.; Larsen, G.L.; Gelfand, E.W. Calcitonin gene-related peptide: Role in airway homeostasis. Curr. Opin. Pharmacol. 2004, 4, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Beltrán, E.; van den Brink, A.M. Understanding CGRP and Cardiovascular Risk. Handb. Exp. Pharmacol. 2019, 255, 131–140. [Google Scholar]
- Favoni, V.; Giani, L.; Al-Hassany, L.; Asioli, G.M.; Butera, C.; de Boer, I.; Guglielmetti, M.; Koniari, C.; Mavridis, T.; Vaikjärv, M.; et al. European Headache Federation School of Advanced Studies (EHF-SAS). CGRP and migraine from a cardiovascular point of view: What do we expect from blocking CGRP? J. Headache Pain. 2019, 20, 27. [Google Scholar] [CrossRef]
- Nahama, A.; Ramachandran, R.; Cisternas, A.F.; Ji, H. The role of afferent pulmonary innervation in poor prognosis of acute respiratory distress syndrome in COVID-19 patients and proposed use of resiniferatoxin (RTX) to improve patient outcomes in advanced disease state: A review. Med. Drug Discov. 2020. [Google Scholar] [CrossRef]
- Johnson, D.S.; Stiff, C.; Lazerwith, S.E.; Kesten, S.R.; Fay, L.K.; Morris, M.; Beidler, D.; Liimatta, M.B.; Smith, S.E.; Dudley, D.T.; et al. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med. Chem. Lett. 2011, 2, 91–96. [Google Scholar] [CrossRef]
- Ahn, K.; Smith, S.E.; Liimatta, M.B.; Beidler, D.; Sadagopan, N.; Dudley, D.T.; Young, T.; Wren, P.; Zhang, Y.; Swaney, S.; et al. Mechanistic and pharmacological characterization of PF-04457845: A highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther. 2011, 338, 114–124. [Google Scholar] [CrossRef]
- D’Souza, D.C.; Cortes-Briones, J.; Creatura, G.; Bluez, G.; Thurnauer, H.; Deaso, E.; Bielen, K.; Surti, T.; Radhakrishnan, R.; Gupta, A.; et al. Efficacy and safety of a fatty acid amide hydrolase inhibitor (PF-04457845) in the treatment of cannabis withdrawal and dependence in men: A double-blind, placebo-controlled, parallel group, phase 2a single-site randomised controlled trial. Lancet Psychiatr. 2019, 6, 35–45. [Google Scholar] [CrossRef]
- Berry, H.; Coquelin, J.P.; Gordon, A.; Seymour, D. Antrafenine, naproxen and placebo in osteoarthritis: A comparative study. Br. J. Rheumatol. 1983, 22, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Palanki, M.S.; Akiyama, H.; Campochiaro, P.; Cao, J.; Chow, C.P.; Dellamary, L.; Doukas, J.; Fine, R.; Gritzen, C.; Hood, J.D.; et al. Development of prodrug 4-chloro-3-(5-methyl-3-{[4-(2-pyrrolidin-1-ylethoxy)phenyl]amino}-1,2,4-benzotriazin-7-yl)phenyl benzoate (TG100801): A topically administered therapeutic candidate in clinical trials for the treatment of age-related macular degeneration. J. Med. Chem. 2008, 51, 1546–1559. [Google Scholar] [PubMed]
- Sharma, S.; Mulik, S.; Kumar, N.; Suryawanshi, A.; Rouse, B.T. An anti-inflammatory role of VEGFR2/Src kinase inhibitor in herpes simplex virus 1-induced immunopathology. J. Virol. 2011, 85, 5995–6007. [Google Scholar] [CrossRef] [Green Version]
- Abidi, A.; Shukla, P.; Ahmad, A. Lifitegrast: A novel drug for treatment of dry eye disease. J. Pharmacol. Pharmacother. 2016, 7, 194–198. [Google Scholar] [CrossRef] [Green Version]
- Paton, D.M. Lifitegrast: First LFA-1/ICAM-1 antagonist for treatment of dry eye disease. Drugs Today 2016, 52, 485–493. [Google Scholar] [CrossRef]
- Xia, J.; Tong, J.; Liu, M.; Shen, Y.; Guo, D. Evaluation of coronavirus in tears and conjunctival secretions of patients with SARS-CoV-2 infection. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. China Medical Treatment Expert Group for Covid-19. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Seah, I.; Agrawal, R. Can the Coronavirus Disease 2019 (COVID-19) Affect the Eyes? A Review of Coronaviruses and Ocular Implications in Humans and Animals. Ocul. Immunol. Inflamm. 2020, 28, 391–395. [Google Scholar] [CrossRef]
- Chen, L.; Deng, C.; Chen, X.; Zhang, X.; Chen, B.; Yu, H.; Qin, Y.; Xiao, K.; Zhnag, H.; Sun, X. Ocular manifestations and clinical characteristics of 534 cases of COVID-19 in China: A cross-sectional study. MedRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, R.F.; McCarron, P.A.; Tunney, M.M. Antifungal photodynamic therapy. Microbiol. Res. 2008, 163, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nikolaeva-Glomb, L.; Mukova, L.; Nikolova, N.; Kussovski, V.; Doumanova, L.; Mantareva, V.; Angelov, I.; Wöhrle, D.; Galabov, A.S. Photodynamic Effect of some Phthalocyanines on Enveloped and Naked Viruses. Acta Virol. 2017, 61, 341–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remichkova, M.; Mukova, L.; Nikolaeva-Glomb, L.; Nikolova, N.; Doumanova, L.; Mantareva, V.; Angelov, I.; Kussovski, V.; Galabov, A.S. Virus inactivation under the photodynamic effect of phthalocyanine zinc(II) complexes. Z. Naturforsch. C J. Biosci. 2017, 72, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Korneev, D.; Kurskaya, O.; Sharshov, K.; Eastwood, J.; Strakhovskaya, M. Ultrastructural Aspects of Photodynamic Inactivation of Highly Pathogenic Avian H5N8 Influenza Virus. Viruses 2019, 11, 955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampf, G.; Todt, D.; Pfaender, S.; Steinmann, E. Persistence of coronaviruses on inanimate surfaces and their inactivation with biocidal agents. J. Hosp. Infect. 2020, 104, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Van Doremalen, N.; Bushmaker, T.; Morris, D.H.; Holbrook, M.G.; Gamble, A.; Williamson, B.N.; Tamin, A.; Harcourt, J.L.; Thornburg, N.J.; Gerber, S.I.; et al. Aerosol and Surface Stability of SARS-CoV-2 as Compared with SARS-CoV-1. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Desai, J.; Gan, H.; Barrow, C.; Jameson, M.; Atkinson, V.; Haydon, A.; Millward, M.; Begbie, S.; Brown, M.; Markman, B.; et al. Phase I, Open-Label, Dose-Escalation/Dose-Expansion Study of Lifirafenib (BGB-283), an RAF Family Kinase Inhibitor, in Patients with Solid Tumors. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Ou, S.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 261–270. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Facchinetti, F.; Friboulet, L. Profile of entrectinib and its potential in the treatment of ROS1-positive NSCLC: Evidence to date. Lung Cancer 2019, 10, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Encinar, J.A.; Menendez, J.A. Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2’-O-Methylation of Viral RNA. Viruses 2020, 12, 525. https://doi.org/10.3390/v12050525
Encinar JA, Menendez JA. Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2’-O-Methylation of Viral RNA. Viruses. 2020; 12(5):525. https://doi.org/10.3390/v12050525
Chicago/Turabian StyleEncinar, José Antonio, and Javier A. Menendez. 2020. "Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2’-O-Methylation of Viral RNA" Viruses 12, no. 5: 525. https://doi.org/10.3390/v12050525
APA StyleEncinar, J. A., & Menendez, J. A. (2020). Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2’-O-Methylation of Viral RNA. Viruses, 12(5), 525. https://doi.org/10.3390/v12050525