Chikungunya Outbreak in the Republic of the Congo, 2019—Epidemiological, Virological and Entomological Findings of a South-North Multidisciplinary Taskforce Investigation


 ,
,  , , ,
, , ,  , ,
, ,  , , , , , , , , , , , , , and add
Show full author list
, , , , , , , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sites
2.2. Study Case Definitions
2.3. Laboratory Diagnostics
2.4. Genomic Analyses
2.5. MinION Sequencing Data Analysis
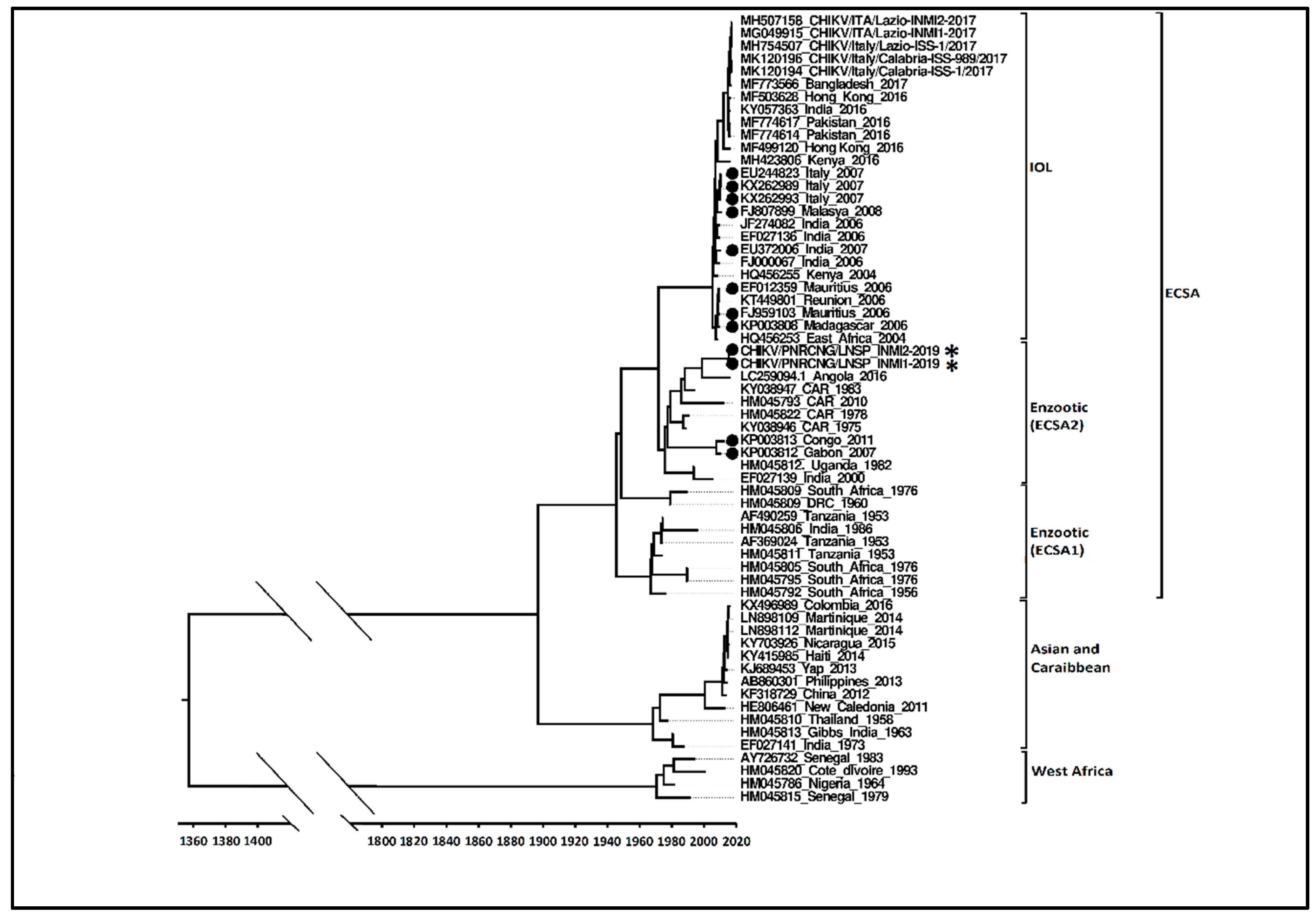
2.6. Phylogenetic Analysis
2.7. Mutational Analysis
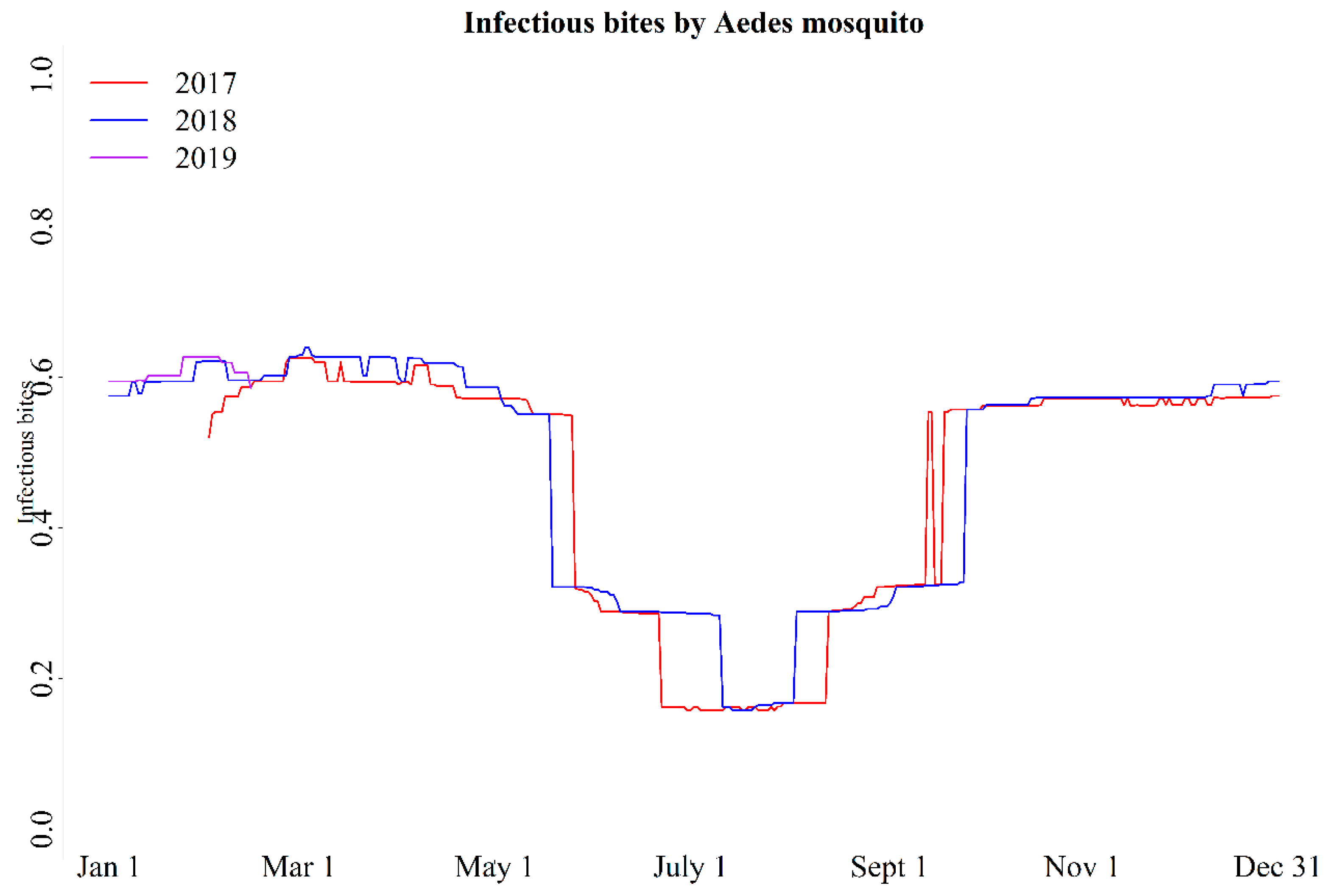
2.8. Estimation of Infectious Bites of CHIKV by Aedes Mosquito
2.9. Statistical Analysis
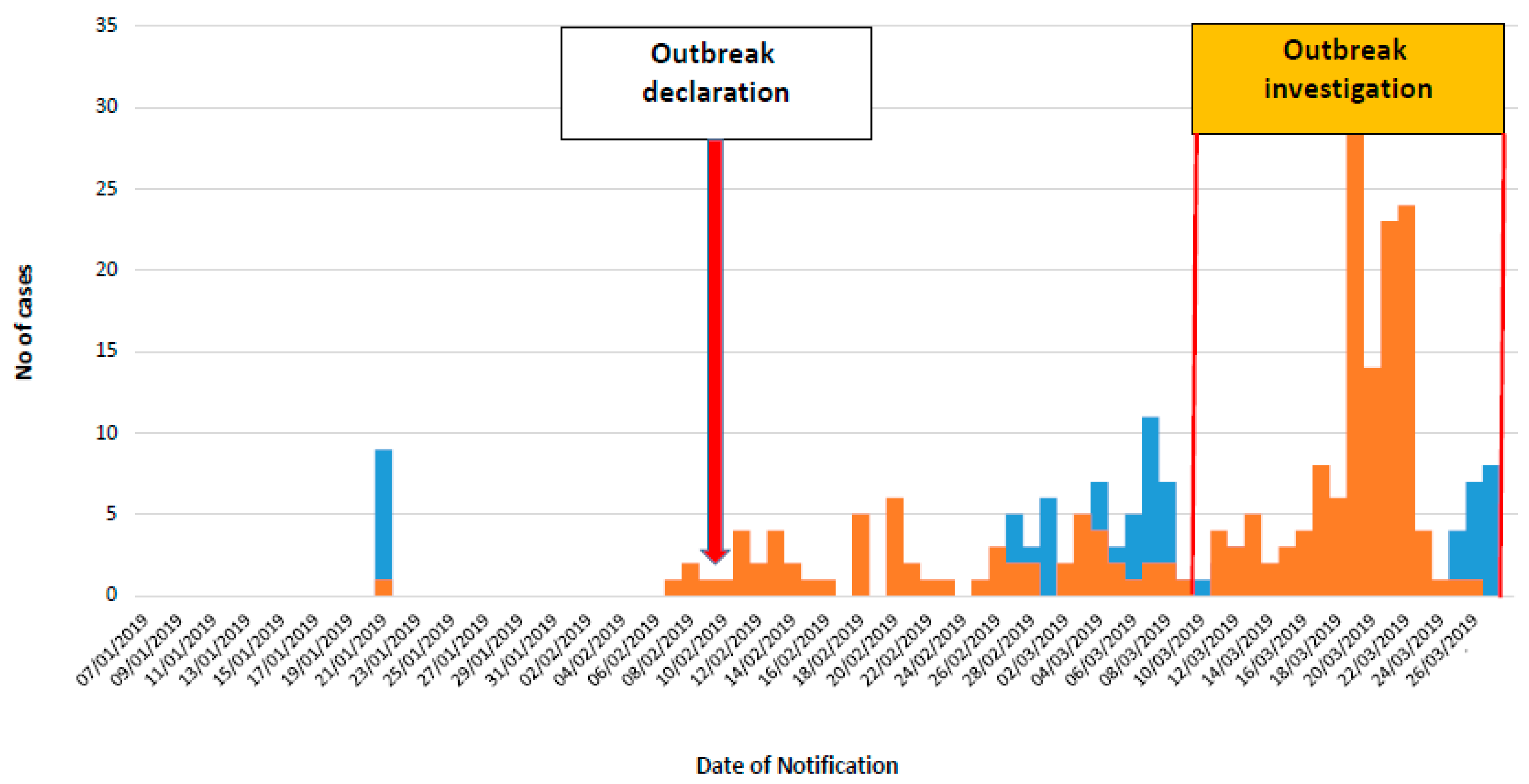
3. Results
3.1. Patients’ Characteristics
3.2. Molecular Characterization
3.3. Predictive Model
3.3.1. Infectious Bites
3.3.2. Entomological Investigations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zumla, A.; Ippolito, G.; McCloskey, B.; Bates, M.; Ansumana, R.; Heymann, D.; Kock, R.; Ntoumi, F. Enhancing preparedness for tackling new epidemic threats. Lancet Respir. Med. 2017, 5, 606–608. [Google Scholar] [CrossRef]
- Ippolito, G.; Lanini, S.; Brouqui, P.; di Caro, A.; Vairo, F.; Abdulla, S.; Fusco, F.M.; Krishna, S.; Capobianchi, M.R.; Kyobe-Bosa, H.; et al. Ebola: Missed opportunities for Europe-Africa research. Lancet Infect. Dis. 2015, 15, 1254–1255. [Google Scholar] [CrossRef]
- Vairo, F.; Ippolito, G.; Ntoumi, F.; Zumla, A. Chikungunya—Epidemiology, Pathogenesis, Clinical Features, Management and Prevention. Infect. Dis. Clin. N. Am. 2019, in press. [Google Scholar]
- Powers, A.M.; Brault, A.C.; Tesh, R.B. Re-emergence of Chikungunya and O’nyong-nyong viruses: Evidence for distinct geographical lineages and distant evolutionary relationships. J. Gen. Virol. 2000, 81, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Volk, S.M.; Chen, R.; Tsetsarkin, K.A.; Adams, A.P.; Garcia, T.I.; Sall, A.A.; Nasar, F.; Schuh, A.J.; Holmes, E.C.; Higgs, S.; et al. Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J. Virol. 2010, 84, 6497–6504. [Google Scholar] [CrossRef] [Green Version]
- Mourya, D.T.; Thakare, J.R.; Gokhale, M.D.; Powers, A.M.; Hundekar, S.L.; Jayakumar, P.C.; Bondre, V.P.; Shouche, Y.S.; Padbidri, V.S. Isolation of chikungunya virus from Aedes aegypti mosquitoes collected in the town of Yawat, Pune District, Maharashtra State. India Acta Virol. 2001, 45, 305–309. [Google Scholar]
- Weaver, S.C.; Forrester, N.L. Chikungunya: Evolutionary history and recent epidemic spread. Antivir. Res. 2015, 120, 32–39. [Google Scholar] [CrossRef]
- Kraemer, M.U.G.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.N.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; van Bortel, W.; et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLife 2015, 4, e08347. [Google Scholar] [CrossRef]
- Peyrefitte, C.N.; Rousset, D.; Pastorino, B.A.M.; Pouillot, R.; Bessaud, M.; Tock, F.; Mansaray, H.; Merle, O.L.; Pascual, A.M.; Paupy, C.; et al. Chikungunya virus, Cameroon, 2006. Emerg. Infect. Dis. 2007, 13, 768–771. [Google Scholar] [CrossRef]
- Leroy, E.M.; Nkoghe, D.; Ollomo, B.; Nze-Nkogue, C.; Becquart, P.; Grard, G.; Pourrut, X.; Charrel, R.; Moureau, G.; Ndjoyi-Mbiguino, A.; et al. Concurrent Chikungunya and dengue virus infections during simultaneous outbreaks, Gabon, 2007. Emerg. Infect. Dis. 2009, 15, 591–593. [Google Scholar] [CrossRef]
- De Lamballerie, X.; Leroy, E.; Charrel, R.N.; Ttsetsarkin, K.; Higgs, S.; Gould, E.A. Chikungunya virus adapts to tiger mosquito via evolutionary convergence: A sign of things to come? Virol. J. 2008, 5, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Moyen, N.; Thiberville, S.; Pastorino, B.; Nougairede, A.; Thirion, L.; Mombouli, J.; Dimi, Y.; Leparc-Goffart, I.; Capobianchi, M.R.; Lepfoundzou, A.D.; et al. First reported chikungunya fever outbreak in the republic of Congo, 2011. PLoS ONE 2014, 9, e115938. [Google Scholar] [CrossRef] [Green Version]
- Mombouli, J.V.; Bitsindou, P.; Elion, D.O.A.; Grolla, A.; Feldmann, H.; Niama, F.R.; Parra, H.; Munster, V.J. Chikungunya virus infection, Brazzaville, Republic of Congo, 2011. Emerg. Infect. Dis. 2013, 19, 1542–1543. [Google Scholar] [CrossRef] [PubMed]
- Vairo, F.; Mammone, A.; Lanini, S.; Nicastri, E.; Castilletti, C.; Carletti, F.; Puro, V.; di Lallo, D.; Panella, V.; Varrenti, D.; et al. Local transmission of chikungunya in Rome and the Lazio region, Italy. PLoS ONE 2018, 13, e0208896. [Google Scholar] [CrossRef]
- Carletti, F.; Marsella, P.; Colavita, F.; Meschi, S.; Lalle, E.; Bordi, L.; di Lallo, D.; Panella, V.; di Caro, A.; Nicastri, E.; et al. Full-Length Genome Sequence of a Chikungunya Virus Isolate from the 2017 Autochthonous Outbreak, Lazio Region, Italy. Genome Announc. 2017, 5, e01306-17. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.H.; Morita, K.; Parquet Md Mdel, C.; Hasebe, F.; Mathenge, E.G.; Igarashi, A. Complete nucleotide sequence of chikungunya virus and evidence for an internal polyadenylation site. J. Gen. Virol. 2002, 83, 3075–3784. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu:scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Haider, N.; Laaksonen, S.; Kjær, L.J.; Oksanen, A.; Bødker, R. The annual, temporal and spatial pattern of Setaria tundra outbreaks in Finnish reindeer: A mechanistic transmission model approach. Parasit Vectors 2018, 11, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, N.; Cuellar, A.C.; Kjær, L.J.; Sørensen, J.H.; Bødker, R. Microclimatic temperatures at Danish cattle farms, 2000-2016: Quantifying the temporal and spatial variation in the transmission potential of Schmallenberg virus. Parasit Vectors 2018, 11, 128. [Google Scholar] [CrossRef] [PubMed]
- Fritz, M.; Taty Taty, R.; Portella, C.; Guimbi, C.; Mankou, M.; Leroy, E.M.; Becquart, P. Re-emergence of chikungunya in the Republic of the Congo in 2019 associated with a possible vector-host switch. Int. J. Infect. Dis. 2019, 84, 99–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaya, S.; Kutsuna, S.; Nakayama, E.; Taniguchi, S.; Tajima, S.; Katanami, Y.; Yamamoto, K.; Takeshita, N.; Hayakawa, K.; Kato, Y.; et al. Chikungunya Fever in Traveler from Angola to Japan, 2016. Emerg. Infect. Dis. 2017, 23, 156–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ISchuffenecker, S.; Iteman, I.; Michault, A.; Murri, S.; Frangeul, L.; Vaney, M.; Lavenir, R.; Pardigon, N.; Reynes, J.; Pettinelli, F.; et al. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 2006, 3, e263. [Google Scholar]
- Tsetsarkin, K.A.; McGee, C.E.; Volk, S.M.; Vanlandingham, D.L.; Weaver, S.C.; Higgs, S. Epistatic roles of E2 glycoprotein mutations in adaption of chikungunya virus to Aedes albopictus and Ae. aegypti mosquitoes. PLoS ONE 2009, 4, e6835. [Google Scholar]
- Agarwal, A.; Sharma, A.K.; Sukumaran, D.; Parida, M.; Dash, P.K. Two novel epistatic mutations (E1:K211E and E2:V264A) in structural proteins of Chikungunya virus enhance fitness in Aedes aegypti. Virology 2016, 497, 59–68. [Google Scholar] [CrossRef]
- Kamgang, B.; Wilson-Bahun, T.A.; Irving, H.; Kusimo, M.O.; Lenga, A.; Wondji, C.S. Geographical distribution of Aedes aegypti and Aedes albopictus (Diptera: Culicidae) and genetic diversity of invading population of Ae. albopictus in the Republic of the Congo. Version 3. Wellcome Open Res. 2018, 3, 79. [Google Scholar] [CrossRef]
- Althouse, B.M.; Guerbois, M.; Cummings, D.A.T.; Diop, O.M.; Faye, O.; Faye, A.; Diallo, D.; Sadio, B.D.; Sow, A.; Faye, O.; et al. Role of monkeys in the sylvatic cycle of chikungunya virus in Senegal. Nat. Commun. 2018, 9, 1046. [Google Scholar] [CrossRef] [Green Version]
- Makiala-Mandanda, S.; Ahuka-Mundeke, S.; Abbate, J.L.; Pukuta-Simbu, E.; Nsio-Mbeta, J.; Berthet, N.; Leroy, E.M.; Becquart, P.; Muyembe-Tamfum, J. Identification of Dengue and Chikungunya Cases Among Suspected Cases of Yellow Fever in the Democratic Republic of the Congo. Vector Borne Zoonotic Dis. 2018, 18, 364–370. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Traits | Source | Ref |
|---|---|---|
| Mosquitoes daily survival rate | e−1/(−4.4 + 1.31*Tmean − 0.03*(Tmean)^2) | [24] |
| Biting rate | (1/(0.0943 + 0.0043*T))/24 | [25] |
| Extrinsic incubation period | (−0.1393 + 0.008T)/24 | [5] |
| Viremic period | 4 days | [26] |
| Mosquitoes to human transmission rate | 0.8 | [26] |
| Human to mosquito transmission rate | 0.8 | [26] |
| Probability of finding a susceptible human host by Aedes spp mosquito | 0.5 | [26] |
| Characteristics * | Total n (%) | Confirmed n (%) | Suspect n (%) | Non Case n (%) | p Value |
|---|---|---|---|---|---|
| Department (N = 349) | 0.217 | ||||
| Pointe–Noire | 288 (100) | 58 (20.1) | 186 (66.6) | 44 (15.3) | |
| Kouilou | 60 (100) | 25 (41.7) | 8 (13.3) | 27 (45.0) | |
| Other | 1 (100) | 1 (100) | 0 | 0 | |
| Gender (N = 302) | 0.217 | ||||
| Male | 113 (100) | 25 (22.1) | 65 (57·5) | 23 (20.4) | |
| Female | 189 (100) | 37 (19.6) | 126 (66·7) | 26 (13.8) | |
| Age (N = 212) | |||||
| Median (IQR) | 34 (13–47) | 41 (30–49) | 34 (13–46) | 33 (13–48) | |
| Age (N = 212) | 0.7 | ||||
| ≤5 years | 4 (100) | 0 | 3 (75.0) | 1 (25.0) | |
| 6–15 years | 60 (100) | 2 (3.3) | 54 (90.0) | 4 (6.7) | |
| 16–40 years | 68 (100) | 4 (5.9) | 59 (86.8) | 5 (7.4) | |
| >40 | 80 (100) | 6 (7.5) | 70 (87.5) | 4 (5.0) | |
| Duration of symptoms (N = 248), Median (IQR) | 3 (2–4) | 3 (2–4) | 2 (1–4) | 3 (2–5) |
| Polyprotein | ORF 1 | ORF 2 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino acid position | 476 | 665 | 680 | 1705 | 1857 | 1918 | 1948 | 2144 | 62 | 404 | 489 | 530 | 589 | 637 | 700 | 711 | 1020 | 1035 | 1078 | 1093 | 1126 | 1131 |
| Protein | nsP1 | nsP2 | nsP3 | nSP4 | C | E2 | E1 | |||||||||||||||
| Amino acid position | 476 | 130 | 145 | 372 | 524 | 55 | 85 | 281 | 62 | 79 | 164 | 205 | 264 | 312 | 375 | 386 | 211 | 226 | 269 | 284 | 317 | 322 |
| Strains non carrying the A226V mutation | ||||||||||||||||||||||
| AF369024 Tanzania 1953 | P | H | E | D | R | S | R | V | R | G | A | G | V | T | S | V | K | A | M | D | I | V |
| LC259094 Angola 2016 | E | T | V | A | ||||||||||||||||||
| MF503628 Hong Kong 2016 | Y | D | N | E | T | A | M | T | A | E | V | E | A | |||||||||
| EF027136 India 2006 | E | T | M | T | A | V | E | A | ||||||||||||||
| JF274082 India 2006 | R | E | T | M | T | A | V | E | A | |||||||||||||
| FJ000067 India 2006 | E | T | M | T | A | V | E | A | ||||||||||||||
| EF027139 India 2000 | R | E | T | V | A | |||||||||||||||||
| HM045793 CAR?? | E | T | V | A | ||||||||||||||||||
| AF490259 Tanzania 1953 | R | E | . | |||||||||||||||||||
| HM045813 Gibbs India 1963 | . | E | D | E | A | |||||||||||||||||
| KY703926 Nicaragua 2015 | E | D | E | A | ||||||||||||||||||
| KJ689453 Yap 2013 | E | D | E | A | ||||||||||||||||||
| KY415985 Haiti 2014 | E | D | E | A | ||||||||||||||||||
| LN898109 Martinique 2014 | E | D | E | A | ||||||||||||||||||
| LN898112 Martinique 2014 | E | D | E | A | ||||||||||||||||||
| HM045786 Nigeria 1964 | G | E | V | A | ||||||||||||||||||
| AY726732 Senegal 1983 | G | E | V | A | ||||||||||||||||||
| HM045809 South Africa 1976 | Q | E | V | A | ||||||||||||||||||
| HM045795 South Africa 1976 | E | A | ||||||||||||||||||||
| KY057363 India 2016 | Y | D | N | E | T | A | M | T | A | E | . | V | E | V | A | |||||||
| HM045811 Tanzania 1953 | E | A | ||||||||||||||||||||
| HM045821 Senegal 1963 | R | . | ||||||||||||||||||||
| HM045810 Thailand 1958 | E | D | E | . | A | |||||||||||||||||
| HM045815 Senegal 1979 | G | E | . | V | A | |||||||||||||||||
| HM045820 Cote dIvoire 1993 | G | E | . | V | A | |||||||||||||||||
| AB860301 Philippines 2013 | E | D | E | A | ||||||||||||||||||
| KX496989 Colombia 2016 | E | D | E | A | ||||||||||||||||||
| EF027141 India 1973 | E | D | E | A | ||||||||||||||||||
| HE806461 New Caledonia 2011 | E | D | E | A | ||||||||||||||||||
| KF318729 China 2012 | E | D | E | A | ||||||||||||||||||
| HQ456255 Kenya 2004 | E | T | M | T | A | V | E | A | ||||||||||||||
| MG049915 CHIKV/ITA/Lazio-INMI1-2017 | Y | D | E | N | C | E | T | S | A | M | T | A | E | V | E | V | A | |||||
| MH507158 CHIKV/ITA/Lazio-INMI2-2017 | Y | D | E | N | C | E | T | S | A | M | T | A | E | V | E | V | A | |||||
| MF774617 Pakistan-07/2016 | Q | Y | D | N | I | E | T | A | M | T | A | E | V | E | V | A | ||||||
| MF774614 Pakistan-04/2016 | Y | D | N | I | E | T | A | M | T | A | E | V | E | V | A | |||||||
| MF773566 Bangladesh 2017 | Y | D | E | N | . | E | T | S | A | M | T | A | E | V | E | V | A | |||||
| MF499120 Hong Kong 2016 | Y | E | T | A | M | T | A | E | V | E | V | A | ||||||||||
| HM045805 South Africa 1976 | E | X | A | |||||||||||||||||||
| HM045792 South Africa 1956 | E | A | ||||||||||||||||||||
| MH423806 Kenya 2016 | E | T | A | M | T | A | E | V | E | A | ||||||||||||
| HQ456253 East Africa 2004 | E | T | M | T | A | V | E | A | ||||||||||||||
| HM045809 DRC 1960 | Q | E | V | A | ||||||||||||||||||
| HM045822 CAR 1978 | E | T | V | A | ||||||||||||||||||
| KY038946 CAR 1975 | E | T | V | A | ||||||||||||||||||
| KY038947 CAR 1983 | Q | E | T | V | A | |||||||||||||||||
| HM045812. Uganda 1982 | E | T | V | A | ||||||||||||||||||
| MK120194 CHIKV/Italy/Calabria-ISS-1/2017 | Y | D | E | N | C | E | T | S | A | M | T | A | E | V | E | V | A | |||||
| MK120196 CHIKV/Italy/Calabria-ISS-989/2017 | Y | D | E | N | C | E | T | S | A | M | T | A | E | V | E | V | A | |||||
| MH754507 CHIKV/Italy/Lazio-ISS-1/2017 | Y | D | E | N | C | E | T | S | A | M | T | A | E | V | E | V | A | |||||
| Strains carryng the A226V mutation | ||||||||||||||||||||||
| CHIK/PNRCNG/LNSP_INMI2-2019 | E | T | D | V | V | A | ||||||||||||||||
| CHIK/PNRCNG/LNSP_INMI1-2019 | E | T | D | V | V | A | ||||||||||||||||
| KT449801 Reunion 2006 | E | T | M | T | A | V | V | E | A | |||||||||||||
| EU244823 Italy 2007 | E | T | M | T | A | V | V | E | A | |||||||||||||
| KX262993 Italy 2007 | E | T | M | T | A | V | V | E | A | |||||||||||||
| KX262989 Italy 2007 | R | E | T | M | T | A | V | V | E | A | ||||||||||||
| KP003813 Congo 2011 | E | T | V | V | A | |||||||||||||||||
| KP003812 Gabon 2007 | E | T | V | V | A | |||||||||||||||||
| FJ807899 Malasya 2008 | E | T | M | T | A | V | V | E | A | |||||||||||||
| EU372006 India 2007 | E | T | M | T | A | V | V | E | A | |||||||||||||
| FJ959103 Mauritius 2006 | E | T | M | T | A | V | V | E | A | |||||||||||||
| EF012359 Mauritius 2006 | E | T | M | T | A | V | V | E | A | |||||||||||||
| KP003808 Madagascar 2006 | Q | R | E | T | M | T | A | V | V | E | A | |||||||||||
| Collection Time | Department | Village/Quartier | Identified Species (n) | RT-PCR |
|---|---|---|---|---|
| 1st week of March | Kouilou | Nkoungou | Ae. albopictus (18) | Positive |
| Diosso | Ae. albopictus (37) | Negative | ||
| Mengo | Ae. albopictus (8) | Negative | ||
| Matombi | Ae. albopictus (16) | Negative | ||
| Mabindou | Ae. albopictus (25) | Negative | ||
| 1st week of March | Pointe Noire | Mpita | Ae. albopictus (34) | Negative |
| Ae. aegypti (20) | Negative | |||
| Tchiamba Nzassi | Ae. albopictus (47) | Negative | ||
| Siafoumou | Ae. albopictus (10) | Negative | ||
| 3rd week of March | Kouilou | Diosso | Ae. aegypti (2) | Negative |
| Ae. albopictus (24) | Negative | |||
| Culicinae spp (12) | Negative |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vairo, F.; Aimè Coussoud-Mavoungou, M.P.; Ntoumi, F.; Castilletti, C.; Kitembo, L.; Haider, N.; Carletti, F.; Colavita, F.; Gruber, C.E.M.; Iannetta, M.; et al. Chikungunya Outbreak in the Republic of the Congo, 2019—Epidemiological, Virological and Entomological Findings of a South-North Multidisciplinary Taskforce Investigation. Viruses 2020, 12, 1020. https://doi.org/10.3390/v12091020
Vairo F, Aimè Coussoud-Mavoungou MP, Ntoumi F, Castilletti C, Kitembo L, Haider N, Carletti F, Colavita F, Gruber CEM, Iannetta M, et al. Chikungunya Outbreak in the Republic of the Congo, 2019—Epidemiological, Virological and Entomological Findings of a South-North Multidisciplinary Taskforce Investigation. Viruses. 2020; 12(9):1020. https://doi.org/10.3390/v12091020
Chicago/Turabian StyleVairo, Francesco, Martin Parfait Aimè Coussoud-Mavoungou, Francine Ntoumi, Concetta Castilletti, Lambert Kitembo, Najmul Haider, Fabrizio Carletti, Francesca Colavita, Cesare E. M. Gruber, Marco Iannetta, and et al. 2020. "Chikungunya Outbreak in the Republic of the Congo, 2019—Epidemiological, Virological and Entomological Findings of a South-North Multidisciplinary Taskforce Investigation" Viruses 12, no. 9: 1020. https://doi.org/10.3390/v12091020
APA StyleVairo, F., Aimè Coussoud-Mavoungou, M. P., Ntoumi, F., Castilletti, C., Kitembo, L., Haider, N., Carletti, F., Colavita, F., Gruber, C. E. M., Iannetta, M., Messina, F., Lanini, S., Ulrich Judicaël, B., Giombini, E., Montaldo, C., Portella, C., Diafouka-Diatela, S., Rueca, M., Kock, R., ... on behalf of the Pandora-ID-NET Consortium Chikungunya Outbreak Group Taskforce. (2020). Chikungunya Outbreak in the Republic of the Congo, 2019—Epidemiological, Virological and Entomological Findings of a South-North Multidisciplinary Taskforce Investigation. Viruses, 12(9), 1020. https://doi.org/10.3390/v12091020