
The Role of Congenital Cytomegalovirus Infection in Adverse Birth Outcomes: A Review of the Potential Mechanisms

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
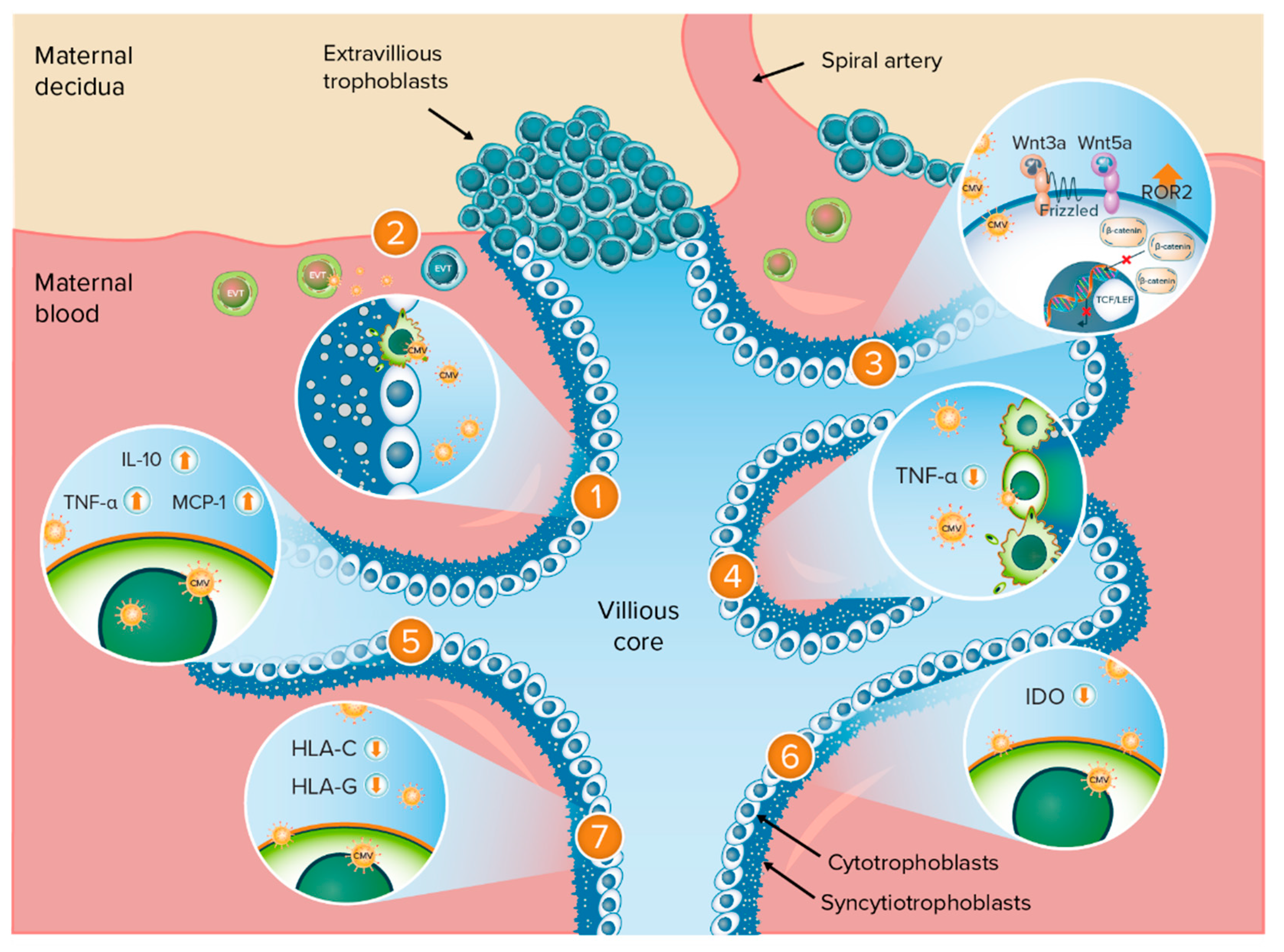
3. Results
3.1. Search Results
3.2. Impairment of TBPC Differentiation and Function
3.3. Impairment of EVT Invasiveness
3.4. Dysregulation of Wnt Signaling Pathways in Cytotrophoblasts
3.5. Trophoblast Apoptosis Mediated by Tumor Necrosis Factor α
3.6. CMV-Induced Cytokine Changes in the Placenta
3.7. Inhibition of Indoleamine 2,3-Dioxygenase Activity
3.8. Downregulation of Trophoblast Class I Major Histocompatibility Complex Molecules
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patro, A.R.K. Subversion of Immune Response by Human Cytomegalovirus. Front. Immunol. 2019, 10, 1155. [Google Scholar] [PubMed] [Green Version]
- Ligat, G.; Cazal, R.; Hantz, S.; Alain, S. The human cytomegalovirus terminase complex as an antiviral target: A close-up view. FEMS Microbiol. Rev. 2018, 42, 137–145. [Google Scholar] [PubMed] [Green Version]
- Mocarski, E.; Shenk, T.; Griffiths, P.; Pass, R.F. Cytomegaloviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1960–2014. [Google Scholar]
- Van Damme, E.; Van Loock, M. Functional annotation of human cytomegalovirus gene products: An update. Front. Microbiol. 2014, 5, 218. [Google Scholar] [PubMed]
- Leruez-Ville, M.; Foulon, I.; Pass, R.; Ville, Y. Cytomegalovirus infection during pregnancy: State of the science. Am. J. Obstet. Gynecol. 2020, 223, 330–349. [Google Scholar] [PubMed]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar]
- Goderis, J.; De Leenheer, E.; Smets, K.; Van Hoecke, H.; Keymeulen, A.; Dhooge, I. Hearing loss and congenital CMV infection: A systematic review. Pediatrics 2014, 134, 972–982. [Google Scholar]
- Duff, P. Diagnosis and management of CMV infection in pregnancy. Perinatology 2010, 1, 1–6. [Google Scholar]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar]
- Benoist, G.; Leruez-Ville, M.; Magny, J.F.; Jacquemard, F.; Salomon, L.J.; Ville, Y. Management of pregnancies with confirmed cytomegalovirus fetal infection. Fetal Diagn. Ther. 2013, 33, 203–214. [Google Scholar]
- Saldan, A.; Forner, G.; Mengoli, C.; Gussetti, N.; Palu, G.; Abate, D. Testing for cytomegalovirus in pregnancy. J. Clin. Microbiol. 2017, 55, 693–702. [Google Scholar] [PubMed] [Green Version]
- Tilburgs, T.; Strominger, J.L. CD8+ effector T cells at the fetal-maternal interface, balancing fetal tolerance and antiviral immunity. Am. J. Reprod. Immunol. 2013, 69, 395–407. [Google Scholar] [PubMed] [Green Version]
- Pereira, L.; Tabata, T.; Petitt, M.; Fang-Hoover, J. Congenital cytomegalovirus infection undermines early development and functions of the human placenta. Placenta 2017, 59 (Suppl. 1), S8–S16. [Google Scholar] [PubMed]
- Hamilton, S.T.; Scott, G.; Naing, Z.; Iwasenko, J.; Hall, B.; Graf, N.; Arbuckle, S.; Craig, M.E.; Rawlinson, W.D. Human cytomegalovirus-induces cytokine changes in the placenta with implications for adverse pregnancy outcomes. PLoS ONE 2012, 7, e52899. [Google Scholar]
- Van Zuylen, W.J.; Ford, C.E.; Wong, D.D.; Rawlinson, W.D. Human cytomegalovirus modulates expression of noncanonical Wnt receptor ROR2 to alter trophoblast migration. J. Virol. 2016, 90, 1108–1115. [Google Scholar]
- Fisher, S.; Genbacev, O.; Maidji, E.; Pereira, L. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: Implications for transmission and pathogenesis. J. Virol. 2000, 74, 6808–6820. [Google Scholar]
- Tabata, T.; Petitt, M.; Zydek, M.; Fang-Hoover, J.; Larocque, N.; Tsuge, M.; Gormley, M.; Kauvar, L.M.; Pereira, L. Human cytomegalovirus infection interferes with the maintenance and differentiation of trophoblast progenitor cells of the human placenta. J. Virol. 2015, 89, 5134–5147. [Google Scholar]
- Lazzarotto, T.; Blázquez-Gamero, D.; Delforge, M.-L.; Foulon, I.; Luck, S.; Modrow, S.; Leruez-Ville, M. Congenital Cytomegalovirus Infection: A Narrative Review of the Issues in Screening and Management From a Panel of European Experts. Front. Pediatrics 2020, 8, 13. [Google Scholar]
- Wilkinson, G.W.; Davison, A.J.; Tomasec, P.; Fielding, C.A.; Aicheler, R.; Murrell, I.; Seirafian, S.; Wang, E.C.Y.; Weekes, M.; Lehner, P.J.; et al. Human cytomegalovirus: Taking the strain. Med. Microbiol. Immunol. 2015, 204, 273–284. [Google Scholar]
- Zydek, M.; Petitt, M.; Fang-Hoover, J.; Adler, B.; Kauvar, L.M.; Pereira, L.; Tabata, T. HCMV infection of human trophoblast progenitor cells of the placenta is neutralized by a human monoclonal antibody to glycoprotein B and not by antibodies to the pentamer complex. Viruses 2014, 6, 1346–1364. [Google Scholar]
- Leghmar, K.; Cenac, N.; Rolland, M.; Martin, H.; Rauwel, B.; Bertrand-Michel, J.; Faouder, P.L.; Bénard, M.; Casper, C.; Davrinche, C.; et al. Cytomegalovirus infection triggers the secretion of the PPARgamma agonists 15-hydroxyeicosatetraenoic acid (15-HETE) and 13-hydroxyoctadecadienoic acid (13-HODE) in human cytotrophoblasts and placental cultures. PLoS ONE 2015, 10, e0132627. [Google Scholar] [CrossRef] [PubMed]
- Rauwel, B.; Mariame, B.; Martin, H.; Nielsen, R.; Allart, S.; Pipy, B.; Mandrup, S.; Devignes, M.D.; Evain-Brion, D.; Fournier, T.; et al. Activation of peroxisome proliferator-activated receptor gamma by human cytomegalovirus for de novo replication impairs migration and invasiveness of cytotrophoblasts from early placentas. J. Virol. 2010, 84, 2946–2954. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Suhua, C.; Juanjuan, C.; Zongzhi, Y.; Juan, X.; Dandan, Z. In vitro study on human cytomegalovirus affecting early pregnancy villous EVT’s invasion function. Virol. J. 2011, 8, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto-Tabata, T.; McDonagh, S.; Chang, H.T.; Fisher, S.; Pereira, L. Human cytomegalovirus interleukin-10 downregulates metalloproteinase activity and impairs endothelial cell migration and placental cytotrophoblast invasiveness in vitro. J. Virol. 2004, 78, 2831–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, M.; Zwezdaryk, K.; Ferris, M.; Shan, B.; Morris, C.A.; Sullivan, D.E. Human cytomegalovirus infection dysregulates the canonical Wnt/beta-catenin signaling pathway. PLoS Pathog. 2012, 8, e1002959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, G.; Hemmings, D.G.; Yurochko, A.D.; Guilbert, L.J. Human cytomegalovirus-caused damage to placental trophoblasts mediated by immediate-early gene-induced tumor necrosis factor-alpha. Am. J. Pathol. 2002, 161, 1371–1381. [Google Scholar] [CrossRef]
- Chan, G.; Guilbert, L.J. Ultraviolet-inactivated human cytomegalovirus induces placental syncytiotrophoblast apoptosis in a Toll-like receptor-2 and tumour necrosis factor-alpha dependent manner. J. Pathol. 2006, 210, 111–120. [Google Scholar] [CrossRef]
- Chaudhuri, S.; Lowen, B.; Chan, G.; Davey, A.; Riddell, M.; Guilbert, L.J. Human cytomegalovirus interacts with toll-like receptor 2 and CD14 on syncytiotrophoblasts to stimulate expression of TNFalpha mRNA and apoptosis. Placenta 2009, 30, 994–1001. [Google Scholar] [CrossRef]
- Lopez, H.; Benard, M.; Saint-Aubert, E.; Baron, M.; Martin, H.; Al Saati, T.; Plantavid, M.; Duga-Neulat, I.; Berrebi, A.; Cristini, C.; et al. Novel model of placental tissue explants infected by cytomegalovirus reveals different permissiveness in early and term placentae and inhibition of indoleamine 2,3-dioxygenase activity. Placenta 2011, 32, 522–530. [Google Scholar] [CrossRef]
- Jun, Y.; Kim, E.; Jin, M.; Sung, H.C.; Han, H.; Geraghty, D.E.; Ahn, K. Human cytomegalovirus gene products US3 and US6 down-regulate trophoblast class I MHC molecules. J. Immunol. 2000, 164, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.; Dobric, N.; Scott, I.C.; Su, L.; Starovic, M.; St-Pierre, B.; Egan, S.E.; Kingdom, J.C.P.; Cross, J.C. The Hand1, Stra13 and Gcm1 transcription factors override FGF signaling to promote terminal differentiation of trophoblast stem cells. Dev. Biol. 2004, 271, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.; Kroener, L.L.; Xu, N.; Wang, E.T.; Banks, A.; Williams, J., III; Goodarzi, M.O.; Chen, Y.-d.I.; Tang, J.; Wang, Y.; et al. Function and hormonal regulation of GATA3 in human first trimester placentation. Biol. Reprod. 2016, 95, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, L.; Maidji, E. Cytomegalovirus infection in the human placenta: Maternal immunity and developmentally regulated receptors on trophoblasts converge. Curr. Top. Microbiol. Immunol. 2008, 325, 383–395. [Google Scholar] [PubMed]
- Weisblum, Y.; Panet, A.; Haimov-Kochman, R.; Wolf, D.G. Models of vertical cytomegalovirus (CMV) transmission and pathogenesis. Semin. Immunopathol. 2014, 36, 615–625. [Google Scholar] [CrossRef]
- Pereira, L.; Maidji, E.; McDonagh, S.; Tabata, T. Insights into viral transmission at the uterine-placental interface. Trends Microbiol. 2005, 13, 164–174. [Google Scholar] [CrossRef]
- Knöfler, M.; Pollheimer, J. Human placental trophoblast invasion and differentiation: A particular focus on Wnt signaling. Front. Genet. 2013, 4, 190. [Google Scholar] [CrossRef] [Green Version]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Sonderegger, S.; Pollheimer, J.; Knofler, M. Wnt signalling in implantation, decidualisation and placental differentiation—Review. Placenta 2010, 31, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Rapacz-Leonard, A.; Dabrowska, M.; Janowski, T. Major histocompatibility complex I mediates immunological tolerance of the trophoblast during pregnancy and may mediate rejection during parturition. Mediat. Inflamm. 2014, 2014, 579279. [Google Scholar]
- Pereira, L.; Petitt, M.; Fong, A.; Tsuge, M.; Tabata, T.; Fang-Hoover, J.; Maidji, E.; Zydek, M.; Zhou, Y.; Inoue, N.; et al. Intrauterine growth restriction caused by underlying congenital cytomegalovirus infection. J. Infect. Dis. 2014, 209, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Kovo, M.; Golan, A. In vitro models using the human placenta to study fetal exposure to drugs. Clin. Med. Reprod. Health 2008, 2, 15–24. [Google Scholar] [CrossRef]
- Vaidya, S.S.; Walsh, S.W.; Gerk, P.M. Application of human placental villous tissue explants to study ABC transporter mediated efflux of 2,4-dinitrophenyl-S-glutathione. Curr. Pharm. Biotechnol. 2011, 12, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Van der Zwan, A.; Van der Meer-Prins, E.M.W.; Van Miert, P.P.M.C.; Van den Heuvel, H.; Anholts, J.D.H.; Roelen, D.L.; Claas, F.H.J.; Heidt, S. Cross-Reactivity of Virus-Specific CD8+ T Cells Against Allogeneic HLA-C: Possible Implications for Pregnancy Outcome. Front. Immunol. 2018, 9, 2880. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.A.; Lockridge, K.M.; Salamat, S.; Tinling, S.P.; Yue, Y.; Zhou, S.S.; Gospe, S.M., Jr.; Britt, W.J.; Tarantal, A.F. Nonhuman primate models of intrauterine cytomegalovirus infection. ILAR J. 2006, 47, 49–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisblum, Y.; Panet, A.; Zakay-Rones, Z.; Haimov-Kochman, R.; Goldman-Wohl, D.; Ariel, I.; Falk, H.; Natanson-Yaron, S.; Goldberg, M.D.; Gilad, R.; et al. Modeling of human cytomegalovirus maternal-fetal transmission in a novel decidual organ culture. J. Virol. 2011, 85, 13204–13213. [Google Scholar] [CrossRef] [Green Version]
- Itell, H.L.; Kaur, A.; Deere, J.D.; Barry, P.A.; Permar, S.R. Rhesus monkeys for a nonhuman primate model of cytomegalovirus infections. Curr. Opin. Virol. 2017, 25, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D. Record Number of Monkeys Being Used in U.S. Research. 2018. Available online: https://www.sciencemag.org/news/2018/11/record-number-monkeys-being-used-us-research (accessed on 1 November 2019).
- Grimm, D. 2020 U.S. Spending Bill Restricts Some Animal Research, Pushes for Lab Animal Retirement. 2019. Available online: https://www.sciencemag.org/news/2019/12/2020-us-spending-bill-restricts-some-animal-research-pushes-lab-animal-retirement (accessed on 18 August 2020).
- Reardon, S. Trump Administration Halts Fetal-Tissue Research by Government Scientists. 2019. Available online: https://www.nature.com/articles/d41586-019-01783-6 (accessed on 18 August 2020).
- Johnson, E.L.; Boggavarapu, S.; Johnson, E.S.; Lal, A.A.; Agrawal, P.; Bhaumik, S.K.; Murali-Krishna, K.; Chakraborty, R. Human cytomegalovirus enhances placental susceptibility and replication of human immunodeficiency virus type 1 (HIV-1), which may facilitate in utero HIV-1 transmission. J. Infect. Dis. 2018, 218, 1464–1473. [Google Scholar] [CrossRef]
- Lindholm, K.; O’Keefe, M. Placental cytomegalovirus infection. Arch. Pathol. Lab. Med. 2019, 143, 639–642. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Search No. | Search Terms |
|---|---|
| Disease area | |
| #1 | Cytomegalovirus [Mesh] |
| #2 | Cytomegalovirus [All fields] |
| #3 | “Congenital cytomegalovirus” [All fields] OR (congenital [All fields] AND cytomegalovirus [All fields]) |
| #4 | “congenital cytomegalovirus infection” [All fields] |
| #5 | “congenital cytomegalovirus disease” [All fields] |
| #6 | “cCMV” [All fields] OR “CMV” [All fields] |
| #7 | #1 OR #2 OR #3 OR #4 OR #5 OR #6 |
| Population | |
| #8 | Stillbirth [Mesh] OR Stillbirth [All fields] OR “Fetal Death” [Mesh] OR “Fetal Death” [All fields] OR “Intrauterine fetal death” [All fields] OR “Intrauterine foetal death” [All fields] OR “IUFD” [All fields] OR “adverse pregnancy outcomes” [All fields] OR “negative pregnancy outcomes” [All fields] OR “negative birth outcomes” [All fields] OR “spontaneous abortion” [All fields] OR “Pregnancy Complications, Infectious/drug therapy” [Majr] |
| #9 | #7 AND #8 |
| #10 | “Premature birth” [Mesh] OR “Premature birth” [All fields] OR “Preterm birth” [All fields] OR “Preterm labor” [All fields] OR “Preterm labour” [All fields] OR “Premature labor” [All fields] OR “Premature labour” [All fields] |
| #11 | #7 AND #10 |
| #12 | “Fetal Growth Retardation” [Mesh] OR “Fetal Growth Retardation” [All fields] OR “Intrauterine growth retardation” [All fields] OR “Intrauterine growth restriction” [All fields] OR “Fetal growth restriction” [All fields] OR “Foetal growth restriction” [All fields] OR “FGR” [All fields] OR “IUGR” [All fields] OR “Infant, Small for Gestational Age” [Mesh] OR “small for gestational age” [All fields] |
| #13 | #7 AND #12 |
| Exclusion terms | |
| #14 | “Animals” [Mesh] NOT “Humans” [Mesh] |
| #15 | “Comment” [Publication Type] OR “Letter” [Publication Type] OR “Editorial” [Publication Type] OR “Case Reports” [Publication Type] OR “Clinical Trial, Phase I” [Publication Type] OR “case study” [Title] OR “case studies” [Title] OR “case report” [Title] OR “case reports” [Title] OR “case series” [Title] |
| Relevant studies | |
| #16 | (#9 OR #11 OR #13) NOT (#14 OR #15) |
| Author | Placental Model | CMV Strains |
|---|---|---|
| Studies assessing TBPC differentiation and development | ||
| Tabata et al. [18] |
|
|
| Zydek et al. [21] |
|
|
| Studies assessing impairment of EVT invasiveness | ||
| Fisher et al. [17] |
|
|
| Leghmar et al. [22] |
|
|
| Rauwel et al. [23] |
|
|
| Tao et al. [24] |
|
|
| Yamamoto-Tabata et al. [25] |
|
|
| Studies assessing Wnt signaling | ||
| Angelova et al. [26] |
|
|
| van Zuylen et al. [16] |
|
|
| Apoptosis studies | ||
| Chan et al. [27] |
|
|
| Chan and Guilbert [28] |
|
|
| Chaudhuri et al. [29] |
|
|
| Study assessing cytokine changes | ||
| Hamilton et al. [15] |
|
|
| IDO activity study | ||
| Lopez et al. [30] |
|
|
| MHC study | ||
| Jun et al. [31] |
|
|
| Protein | Function | Impact of CMV Infection of TBPCs on Protein Expression or Localization |
|---|---|---|
| Geminin |
| Upregulated |
| HMGA2 |
| Downregulated a |
| SOX2 |
| Downregulated a |
| GATA3 |
| Altered subcellular localization b:
|
| GATA4 |
| Downregulated |
| Hand1 |
| Altered subcellular localization b:
|
| PPARγ |
| Upregulated |
| hCG |
| Downregulated c |
| Protein | Function | Impact of CMV Infection |
|---|---|---|
| MMP-2 and MMP-9 |
|
|
| c-erbB-2 |
|
|
| CMV IL-10 and human IL-10 |
|
|
| α1β1 integrin |
|
|
| PPARγ |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Njue, A.; Coyne, C.; Margulis, A.V.; Wang, D.; Marks, M.A.; Russell, K.; Das, R.; Sinha, A. The Role of Congenital Cytomegalovirus Infection in Adverse Birth Outcomes: A Review of the Potential Mechanisms. Viruses 2021, 13, 20. https://doi.org/10.3390/v13010020
Njue A, Coyne C, Margulis AV, Wang D, Marks MA, Russell K, Das R, Sinha A. The Role of Congenital Cytomegalovirus Infection in Adverse Birth Outcomes: A Review of the Potential Mechanisms. Viruses. 2021; 13(1):20. https://doi.org/10.3390/v13010020
Chicago/Turabian StyleNjue, Annete, Carolyn Coyne, Andrea V. Margulis, Dai Wang, Morgan A. Marks, Kevin Russell, Rituparna Das, and Anushua Sinha. 2021. "The Role of Congenital Cytomegalovirus Infection in Adverse Birth Outcomes: A Review of the Potential Mechanisms" Viruses 13, no. 1: 20. https://doi.org/10.3390/v13010020
APA StyleNjue, A., Coyne, C., Margulis, A. V., Wang, D., Marks, M. A., Russell, K., Das, R., & Sinha, A. (2021). The Role of Congenital Cytomegalovirus Infection in Adverse Birth Outcomes: A Review of the Potential Mechanisms. Viruses, 13(1), 20. https://doi.org/10.3390/v13010020