
Editing of the TRIM5 Gene Decreases the Permissiveness of Human T Lymphocytic Cells to HIV-1

 , and
, and
Abstract
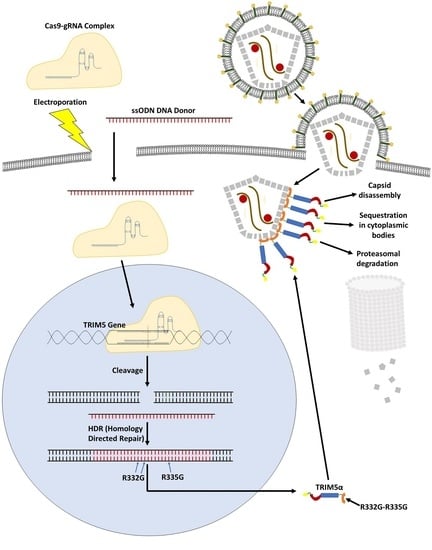
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. TRIM5 Editing
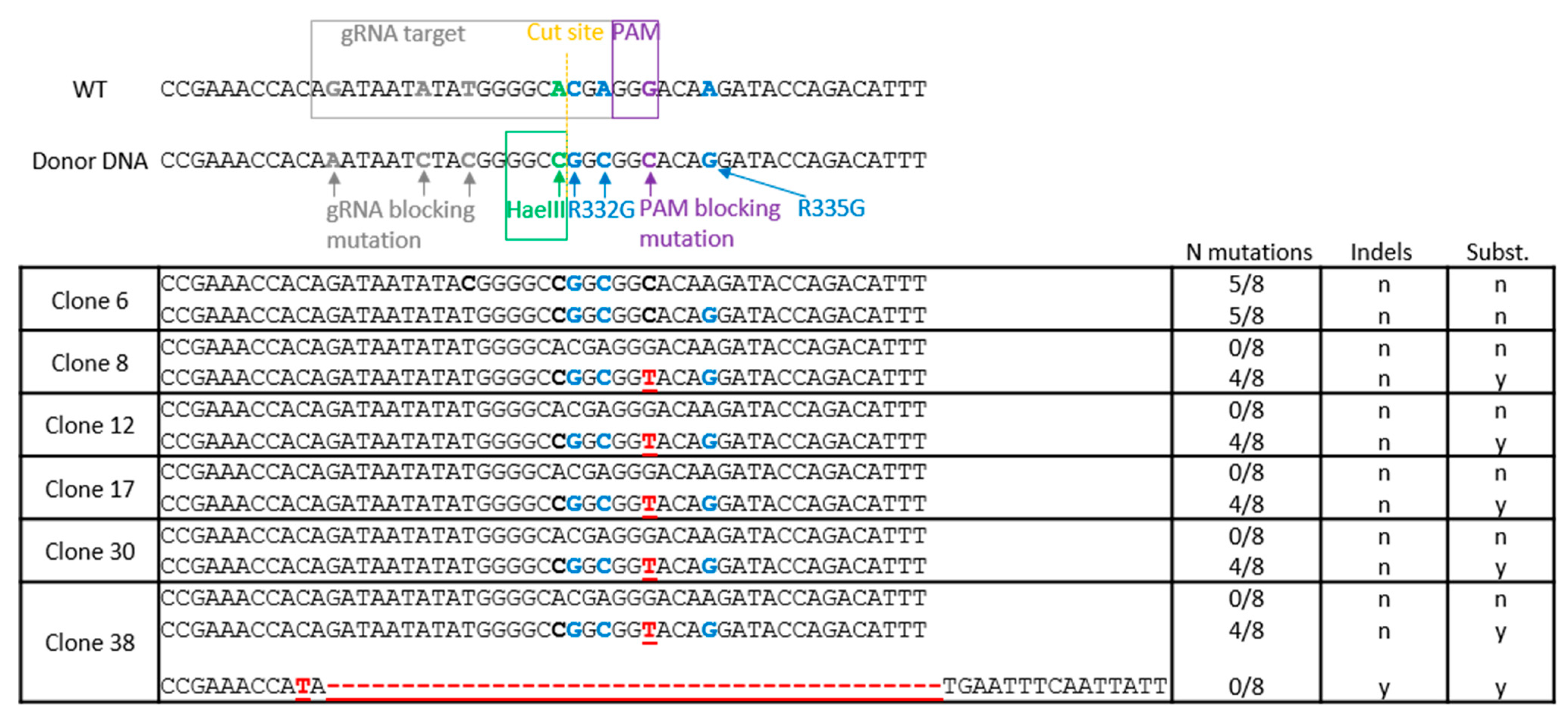
2.3. Isolation and Screening of Cell Clones
2.4. HaeIII Screening of Edited Clones and Deep Sequencing
2.5. Virus Production
2.6. Viral Challenges
2.7. Knockdowns
2.8. HIV-1 cDNA Quantification
3. Results
3.1. TRIM5 Editing
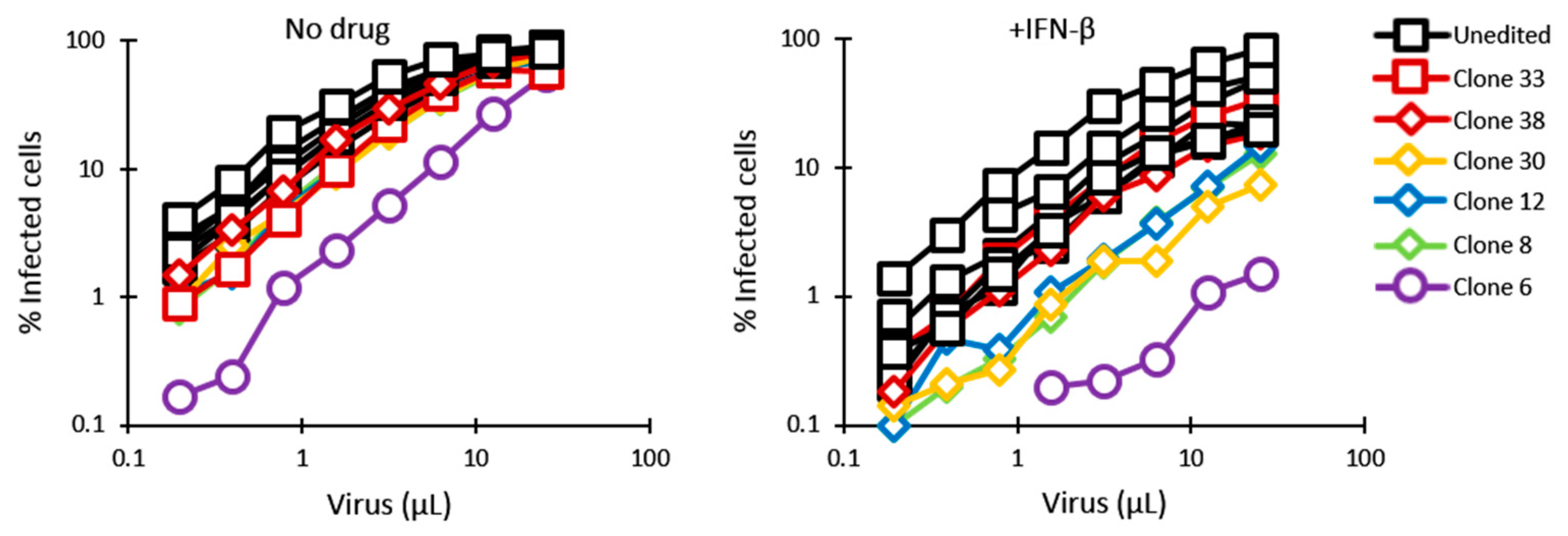
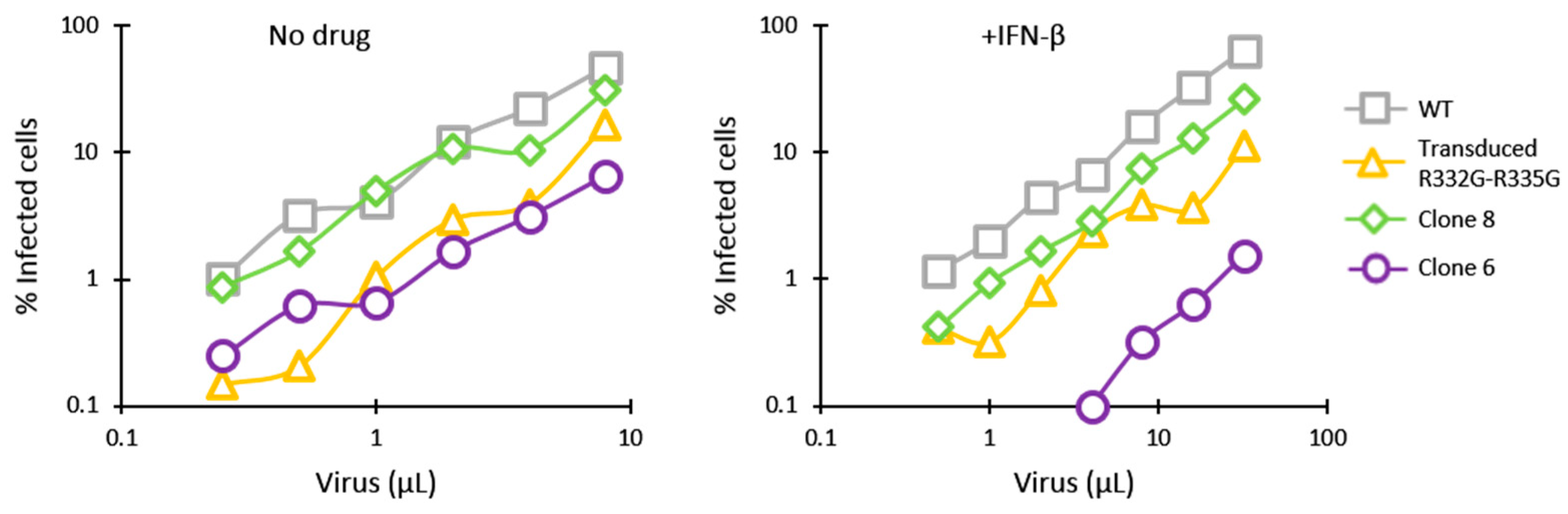
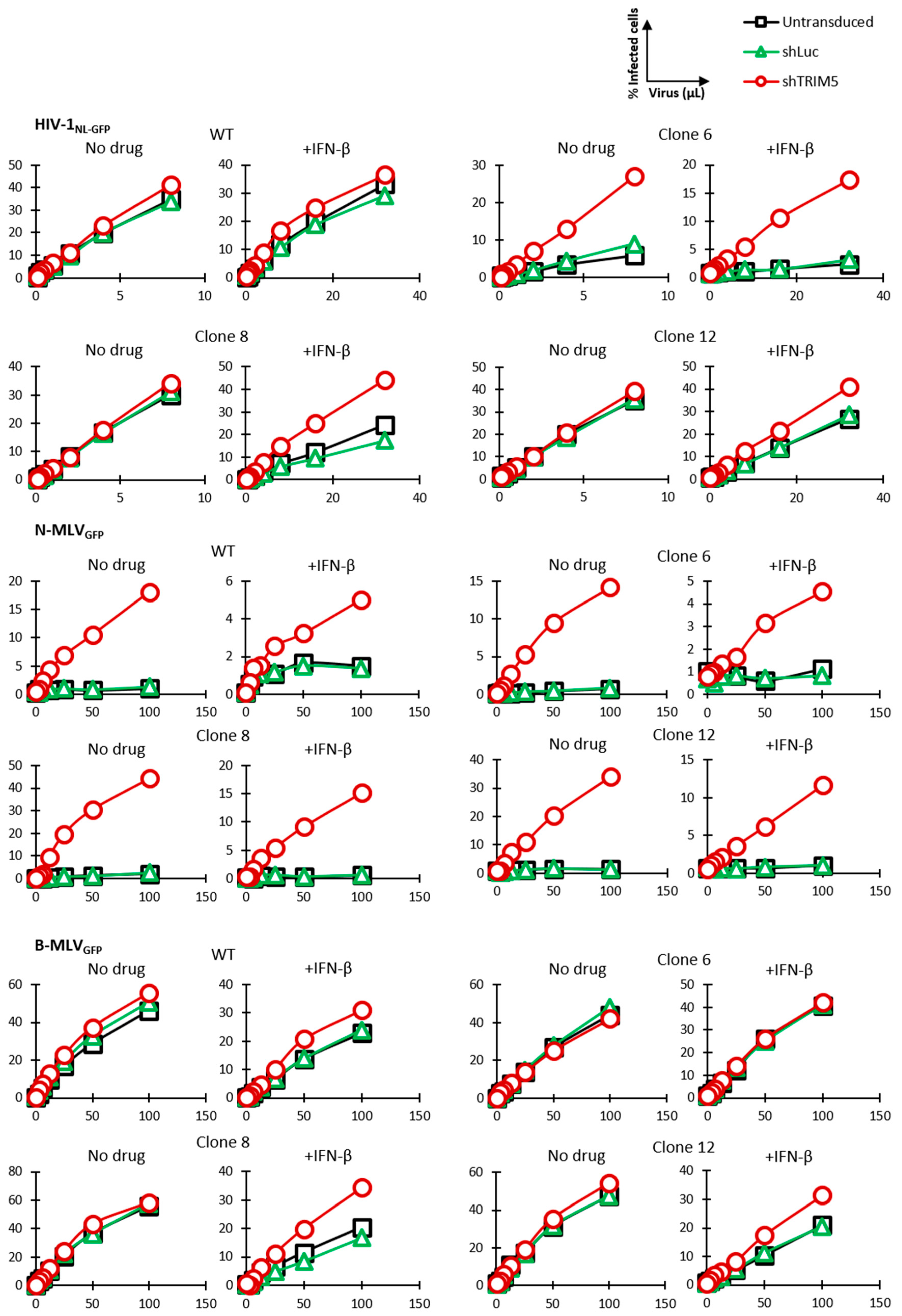
3.2. HIV-1 Restriction Activity in TRIM5-Edited Clones
3.3. Knockdown Validation of TRIM5α Antiviral Function
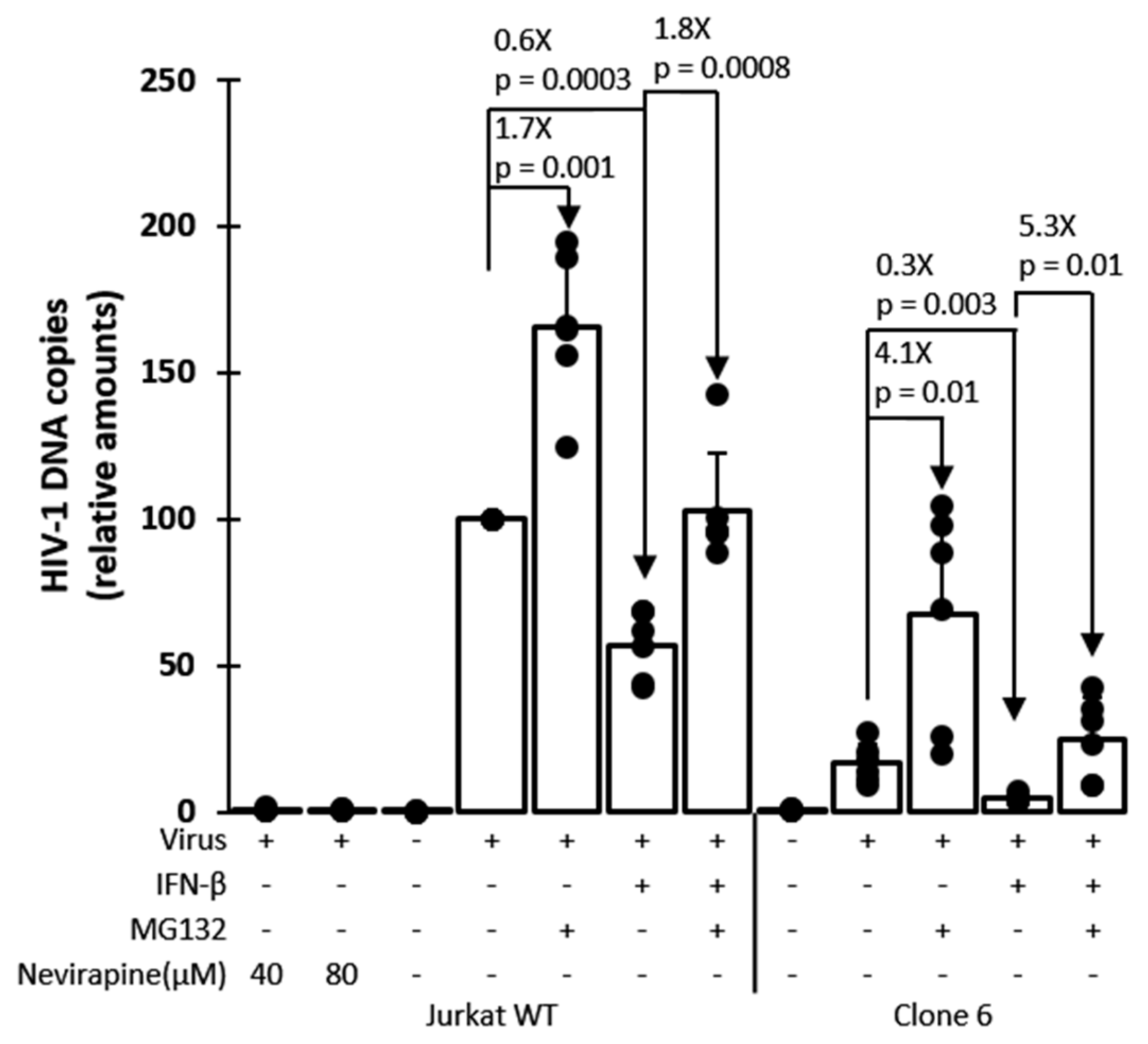
3.4. Mechanism of Inhibition by Edited TRIM5α
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Angel, J.B.; Parato, K.G.; Kumar, A.; Kravcik, S.; Badley, A.D.; Fex, C.; Ashby, D.; Sun, E.; Cameron, D.W. Progressive human immunodeficiency virus-specific immune recovery with prolonged viral suppression. J. Infect. Dis. 2001, 183, 546–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagos, L.; Fessehaye, S.; Anand, I.S. Nature and prevalence of adverse drug reaction of antiretroviral medications in Halibet National Referral Hospital: A retrospective study. BMC Pharmacol. Toxicol. 2019, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef]
- Rose, R.; Nolan, D.J.; Maidji, E.; Stoddart, C.A.; Singer, E.J.; Lamers, S.L.; McGrath, M.S. Eradication of HIV from Tissue Reservoirs: Challenges for the Cure. Aids Res. Hum. Retrovir. 2018, 34, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Yang, H.; Gao, Y.; Chen, Z.; Xie, L.; Liu, Y.; Liu, Y.; Wang, X.; Li, H.; Lai, W.; et al. CRISPR/Cas9-Mediated CCR5 Ablation in Human Hematopoietic Stem/Progenitor Cells Confers HIV-1 Resistance In Vivo. Mol. Ther. 2017, 25, 1782–1789. [Google Scholar] [CrossRef]
- Kang, H.; Minder, P.; Park, M.A.; Mesquitta, W.T.; Torbett, B.E.; Slukvin, I.I. CCR5 Disruption in Induced Pluripotent Stem Cells Using CRISPR/Cas9 Provides Selective Resistance of Immune Cells to CCR5-tropic HIV-1 Virus. Mol. Ther. Nucleic Acids 2015, 4, e268. [Google Scholar] [CrossRef]
- Qi, C.; Li, D.; Jiang, X.; Jia, X.; Lu, L.; Wang, Y.; Sun, J.; Shao, Y.; Wei, M. Inducing CCR5Delta32/Delta32 Homozygotes in the Human Jurkat CD4+ Cell Line and Primary CD4+ Cells by CRISPR-Cas9 Genome-Editing Technology. Mol. Ther. Nucleic Acids 2018, 12, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Verheyen, J.; Thielen, A.; Lubke, N.; Dirks, M.; Widera, M.; Dittmer, U.; Kordales, L.; Daumer, M.; Jong, T.C.M.; Wensing, A.M.J.; et al. Rapid rebound of a preexisting CXCR4-tropic HIV variant after allogeneic transplantation with CCR5 delta32 homozygous stem cells. Clin. Infect. Dis. 2018. [Google Scholar] [CrossRef]
- Kordelas, L.; Verheyen, J.; Beelen, D.W.; Horn, P.A.; Heinold, A.; Kaiser, R.; Trenschel, R.; Schadendorf, D.; Dittmer, U.; Esser, S. Shift of HIV tropism in stem-cell transplantation with CCR5 Delta32 mutation. N. Engl. J. Med. 2014, 371, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Merindol, N.; Berthoux, L. Restriction Factors in HIV-1 Disease Progression. Curr. HIV Res. 2015, 13, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Colomer-Lluch, M.; Ruiz, A.; Moris, A.; Prado, J.G. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front. Immunol. 2018, 9, 2876. [Google Scholar] [CrossRef] [Green Version]
- Carthagena, L.; Parise, M.C.; Ringeard, M.; Chelbi-Alix, M.K.; Hazan, U.; Nisole, S. Implication of TRIM alpha and TRIMCyp in interferon-induced anti-retroviral restriction activities. Retrovirology 2008, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Pornillos, O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 2019, 17, 546–556. [Google Scholar] [CrossRef]
- Passerini, L.D.; Keckesova, Z.; Towers, G.J. Retroviral restriction factors Fv1 and TRIM5alpha act independently and can compete for incoming virus before reverse transcription. J. Virol. 2006, 80, 2100–2105. [Google Scholar] [CrossRef] [Green Version]
- Rajsbaum, R.; Garcia-Sastre, A.; Versteeg, G.A. TRIMmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 2014, 426, 1265–1284. [Google Scholar] [CrossRef] [Green Version]
- Kutluay, S.B.; Perez-Caballero, D.; Bieniasz, P.D. Fates of Retroviral Core Components during Unrestricted and TRIM5-Restricted Infection. Plos Pathog. 2013, 9, e1003214. [Google Scholar] [CrossRef] [Green Version]
- Rold, C.J.; Aiken, C. Proteasomal degradation of TRIM5alpha during retrovirus restriction. PLoS Pathog. 2008, 4, e1000074. [Google Scholar] [CrossRef] [Green Version]
- Imam, S.; Komurlu, S.; Mattick, J.; Selyutina, A.; Talley, S.; Eddins, A.; Diaz-Griffero, F.; Campbell, E.M. K63-Linked Ubiquitin Is Required for Restriction of HIV-1 Reverse Transcription and Capsid Destabilization by Rhesus TRIM5alpha. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, A.J.; Christensen, D.E.; Nelson, C.; Tan, C.P.; Schaller, T.; Lehner, P.J.; Sundquist, W.I.; Towers, G.J. TRIM5alpha requires Ube2W to anchor Lys63-linked ubiquitin chains and restrict reverse transcription. EMBO J. 2015, 34, 2078–2095. [Google Scholar] [CrossRef]
- Merindol, N.; El-Far, M.; Sylla, M.; Masroori, N.; Dufour, C.; Li, J.X.; Cherry, P.; Plourde, M.B.; Tremblay, C.; Berthoux, L. HIV-1 capsids from B27/B57+ elite controllers escape Mx2 but are targeted by TRIM5alpha, leading to the induction of an antiviral state. PLoS Pathog. 2018, 14, e1007398. [Google Scholar] [CrossRef]
- Nepveu-Traversy, M.E.; Berthoux, L. The conserved sumoylation consensus site in TRIM5alpha modulates its immune activation functions. Virus Res. 2014, 184C, 30–38. [Google Scholar] [CrossRef]
- Na, L.; Tang, Y.D.; Wang, C.; Liu, C.; Wang, X. Rhesus monkey TRIM5alpha protein SPRY domain contributes to AP-1 activation. J. Biol. Chem. 2018, 293, 2661–2674. [Google Scholar] [CrossRef] [Green Version]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Stremlau, M.; Perron, M.; Welikala, S.; Sodroski, J. Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J. Virol. 2005, 79, 3139–3145. [Google Scholar] [CrossRef] [Green Version]
- Perez-Caballero, D.; Hatziioannou, T.; Yang, A.; Cowan, S.; Bieniasz, P.D. Human tripartite motif 5alpha domains responsible for retrovirus restriction activity and specificity. J. Virol. 2005, 79, 8969–8978. [Google Scholar] [CrossRef] [Green Version]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting TRIM5alpha protein. Proc. Natl. Acad. Sci. USA 2011, 108, 534–539. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.; Skorupka, K.A.; Pak, A.J.; Ganser-Pornillos, B.K.; Pornillos, O.; Voth, G.A. TRIM5alpha self-assembly and compartmentalization of the HIV-1 viral capsid. Nat. Commun. 2020, 11, 1307. [Google Scholar] [CrossRef]
- Li, X.; Yeung, D.F.; Fiegen, A.M.; Sodroski, J. Determinants of the higher order association of the restriction factor TRIM5alpha and other tripartite motif (TRIM) proteins. J. Biol. Chem. 2011, 286, 27959–27970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keown, J.R.; Yang, J.X.; Douglas, J.; Goldstone, D.C. Characterisation of assembly and ubiquitylation by the RBCC motif of Trim5alpha. Sci. Rep. 2016, 6, 26837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, L.R.; Aiken, C. TRIM5alpha disrupts the structure of assembled HIV-1 capsid complexes in vitro. J. Virol. 2010, 84, 6564–6569. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Zhang, P. CryoEM analysis of capsid assembly and structural changes upon interactions with a host restriction factor, TRIM5alpha. Methods Mol. Biol 2014, 1087, 13–28. [Google Scholar]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastri, J.; Campbell, E.M. Recent insights into the mechanism and consequences of TRIM5alpha retroviral restriction. Aids Res. Hum. Retrovir. 2011, 27, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roa, A.; Hayashi, F.; Yang, Y.; Lienlaf, M.; Zhou, J.; Shi, J.; Watanabe, S.; Kigawa, T.; Yokoyama, S.; Aiken, C.; et al. RING domain mutations uncouple TRIM5alpha restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J. Virol. 2012, 86, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Anderson, J.L.; Campbell, E.M.; Joseph, A.M.; Hope, T.J. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. USA 2006, 103, 7465–7470. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.M.; Weingart, J.; Sette, P.; Opp, S.; Sastri, J.; O’Connor, S.K.; Talley, S.; Diaz-Griffero, F.; Hirsch, V.; Bouamr, F. TRIM5alpha-Mediated Ubiquitin Chain Conjugation Is Required for Inhibition of HIV-1 Reverse Transcription and Capsid Destabilization. J. Virol. 2016, 90, 1849–1857. [Google Scholar] [CrossRef] [Green Version]
- Nepveu-Traversy, M.E.; Demogines, A.; Fricke, T.; Plourde, M.B.; Riopel, K.; Veillette, M.; Diaz-Griffero, F.; Sawyer, S.L.; Berthoux, L. A putative SUMO interacting motif in the B30.2/SPRY domain of rhesus macaque TRIM5alpha important for NF-kappaB/AP-1 signaling and HIV-1 restriction. Heliyon 2016, 2, e00056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, E.M.; Perez, O.; Anderson, J.L.; Hope, T.J. Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5alpha. J. Cell Biol. 2008, 180, 549–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, S.L.; Wu, L.I.; Emerman, M.; Malik, H.S. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. USA 2005, 102, 2832–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatziioannou, T.; Perez-Caballero, D.; Yang, A.; Cowan, S.; Bieniasz, P.D. Retrovirus resistance factors Ref1 and Lv1 are species-specific variants of TRIM5alpha. Proc. Natl. Acad. Sci. USA 2004, 101, 10774–10779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bérubé, J.; Bouchard, A.; Berthoux, L. Both TRIM5alpha and TRIMCyp have only weak antiviral activity in canine D17 cells. Retrovirology 2007, 4, 68. [Google Scholar] [CrossRef] [Green Version]
- Granier, C.; Battivelli, E.; Lecuroux, C.; Venet, A.; Lambotte, O.; Schmitt-Boulanger, M.; Delaugerre, C.; Molina, J.M.; Chakrabarti, L.A.; Clavel, F.; et al. Pressure from TRIM5alpha contributes to control of HIV-1 replication by individuals expressing protective HLA-B alleles. J. Virol. 2013, 87, 10368–10380. [Google Scholar] [CrossRef] [Green Version]
- Battivelli, E.; Migraine, J.; Lecossier, D.; Yeni, P.; Clavel, F.; Hance, A.J. Gag cytotoxic T lymphocyte escape mutations can increase sensitivity of HIV-1 to human TRIM5alpha, linking intrinsic and acquired immunity. J. Virol. 2011, 85, 11846–11854. [Google Scholar] [CrossRef] [Green Version]
- Song, B.; Javanbakht, H.; Perron, M.; Park, D.H.; Stremlau, M.; Sodroski, J. Retrovirus restriction by TRIM5alpha variants from Old World and New World primates. J. Virol. 2005, 79, 3930–3937. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.J.; Webb, B.L.; Maplanka, C.; Newman, R.M.; Verschoor, E.J.; Heeney, J.L.; Towers, G.J. Rhesus macaque TRIM5 alleles have divergent antiretroviral specificities. J. Virol. 2008, 82, 7243–7247. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.E.; Chen, R.X.; McGee, J.; Nacey, C.; Pollard, R.B.; Abedi, M.; Bauer, G.; Nolta, J.A.; Anderson, J.S. Generation of an HIV-1-resistant immune system with CD34(+) hematopoietic stem cells transduced with a triple-combination anti-HIV lentiviral vector. J. Virol. 2012, 86, 5719–5729. [Google Scholar] [CrossRef] [Green Version]
- Yap, M.W.; Nisole, S.; Stoye, J.P. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr. Biol. 2005, 15, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, X.; Stremlau, M.; Lee, M.; Sodroski, J. Removal of arginine 332 allows human TRIM5alpha to bind human immunodeficiency virus capsids and to restrict infection. J. Virol. 2006, 80, 6738–6744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, Q.T.; Bouchard, A.; Grutter, M.G.; Berthoux, L. Generation of human TRIM5alpha mutants with high HIV-1 restriction activity. Gene Ther. 2010, 17, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, Q.T.; Veillette, M.; Brandariz-Nunez, A.; Pawlica, P.; Thibert-Lefebvre, C.; Chandonnet, N.; Diaz-Griffero, F.; Berthoux, L. A novel aminoacid determinant of HIV-1 restriction in the TRIM5alpha variable 1 region isolated in a random mutagenic screen. Virus Res. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, U.; Urak, K.; Veillette, M.; Nepveu-Traversy, M.E.; Pham, Q.T.; Hamel, S.; Rossi, J.J.; Berthoux, L. Preclinical Assessment of Mutant Human TRIM5alpha as an Anti-HIV-1 Transgene. Hum. Gene Ther. 2015, 26, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, C.M.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Tigchelaar, W.; van der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B. Receptor usage dictates HIV-1 restriction by human TRIM5alpha in dendritic cell subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef]
- Battivelli, E.; Migraine, J.; Lecossier, D.; Matsuoka, S.; Perez-Bercoff, D.; Saragosti, S.; Clavel, F.; Hance, A.J. Modulation of TRIM5alpha activity in human cells by alternatively spliced TRIM5 isoforms. J. Virol. 2011, 85, 7828–7835. [Google Scholar] [CrossRef] [Green Version]
- Berthoux, L.; Sebastian, S.; Sayah, D.M.; Luban, J. Disruption of human TRIM5alpha antiviral activity by nonhuman primate orthologues. J. Virol. 2005, 79, 7883–7888. [Google Scholar] [CrossRef] [Green Version]
- Kan, Y.; Ruis, B.; Takasugi, T.; Hendrickson, E.A. Mechanisms of precise genome editing using oligonucleotide donors. Genome Res. 2017, 27, 1099–1111. [Google Scholar] [CrossRef] [Green Version]
- Dufour, C.; Claudel, A.; Joubarne, N.; Merindol, N.; Maisonnet, T.; Masroori, N.; Plourde, M.B.; Berthoux, L. Editing of the human TRIM5 gene to introduce mutations with the potential to inhibit HIV-1. PLoS ONE 2018, 13, e0191709. [Google Scholar] [CrossRef] [Green Version]
- Berthoux, L.; Sebastian, S.; Sokolskaja, E.; Luban, J. Cyclophilin A is required for TRIM5{alpha}-mediated resistance to HIV-1 in Old World monkey cells. Proc. Natl. Acad. Sci. USA 2005, 102, 14849–14853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masroori, N.; Merindol, N.; Berthoux, L. The interferon-induced antiviral protein PML (TRIM19) promotes the restriction and transcriptional silencing of lentiviruses in a context-specific, isoform-specific fashion. Retrovirology 2016, 13, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malbec, M.; Pham, Q.T.; Plourde, M.B.; Letourneau-Hogan, A.; Nepveu-Traversy, M.E.; Berthoux, L. Murine double minute 2 as a modulator of retroviral restrictions mediated by TRIM5alpha. Virology 2010, 405, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Veillette, M.; Bichel, K.; Pawlica, P.; Freund, S.M.; Plourde, M.B.; Pham, Q.T.; Reyes-Moreno, C.; James, L.C.; Berthoux, L. The V86M mutation in HIV-1 capsid confers resistance to TRIM5alpha by abrogation of cyclophilin A-dependent restriction and enhancement of viral nuclear import. Retrovirology 2013, 10, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Chen, Y.; Farzan, M.; Choe, H.; Ohagen, A.; Gartner, S.; Busciglio, J.; Yang, X.; Hofmann, W.; Newman, W.; et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 1997, 385, 645–649. [Google Scholar] [CrossRef]
- Anderson, J.L.; Campbell, E.M.; Wu, X.; Vandegraaff, N.; Engelman, A.; Hope, T.J. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J. Virol. 2006, 80, 9754–9760. [Google Scholar] [CrossRef] [Green Version]
- Margolis, D.M.; Archin, N.M.; Cohen, M.S.; Eron, J.J.; Ferrari, G.; Garcia, J.V.; Gay, C.L.; Goonetilleke, N.; Joseph, S.B.; Swanstrom, R.; et al. Curing HIV: Seeking to Target and Clear Persistent Infection. Cell 2020, 181, 189–206. [Google Scholar] [CrossRef]
- Thomas, J.; Ruggiero, A.; Paxton, W.A.; Pollakis, G. Measuring the Success of HIV-1 Cure Strategies. Front. Cell. Infect. Microbiol. 2020, 10, 134. [Google Scholar] [CrossRef]
- Stephenson, K.E.; Wagh, K.; Korber, B.; Barouch, D.H. Vaccines and Broadly Neutralizing Antibodies for HIV-1 Prevention. Annu. Rev. Immunol. 2020, 38, 673–703. [Google Scholar] [CrossRef]
- Del Moral-Sanchez, I.; Sliepen, K. Strategies for inducing effective neutralizing antibody responses against HIV-1. Expert Rev. Vaccines 2019, 18, 1127–1143. [Google Scholar] [CrossRef] [Green Version]
- Swindells, S.; Andrade-Villanueva, J.F.; Richmond, G.J.; Rizzardini, G.; Baumgarten, A.; Masia, M.; Latiff, G.; Pokrovsky, V.; Bredeek, F.; Smith, G.; et al. Long-Acting Cabotegravir and Rilpivirine for Maintenance of HIV-1 Suppression. N. Engl. J. Med. 2020, 382, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Bester, S.M.; Wei, G.; Zhao, H.; Adu-Ampratwum, D.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Annamalai, A.S.; Singh, P.K.; Shkriabai, N.; et al. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science 2020, 370, 360–364. [Google Scholar] [PubMed]
- Blair, H.A. Ibalizumab: A Review in Multidrug-Resistant HIV-1 Infection. Drugs 2020, 80, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Katz, I.T.; Maughan-Brown, B. Improved life expectancy of people living with HIV: Who is left behind? Lancet HIV 2017, 4, e324–e326. [Google Scholar] [CrossRef] [Green Version]
- Basavaraj, K.H.; Navya, M.A.; Rashmi, R. Quality of life in HIV/AIDS. Indian J. Sex. Transm. Dis. Aids 2010, 31, 75–80. [Google Scholar] [CrossRef]
- Pozniak, A. Quality of life in chronic HIV infection. Lancet HIV 2014, 1, e6–e7. [Google Scholar] [CrossRef] [Green Version]
- Koujah, L.; Shukla, D.; Naqvi, A.R. CRISPR-Cas based targeting of host and viral genes as an antiviral strategy. Semin. Cell Dev. Biol. 2019, 96, 53–64. [Google Scholar] [CrossRef]
- Gaj, T.; Staahl, B.T.; Rodrigues, G.M.C.; Limsirichai, P.; Ekman, F.K.; Doudna, J.A.; Schaffer, D.V. Targeted gene knock-in by homology-directed genome editing using Cas9 ribonucleoprotein and AAV donor delivery. Nucleic Acids Res. 2017, 45, e98. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhang, X.; Wang, H.; Liu, D.; Li, Z.; Wu, Z.; Yang, H. Increasing CRISPR/Cas9-mediated homology-directed DNA repair by histone deacetylase inhibitors. Int. J. Biochem. Cell Biol. 2020, 125, 105790. [Google Scholar] [CrossRef]
- Guo, Q.; Mintier, G.; Ma-Edmonds, M.; Storton, D.; Wang, X.; Xiao, X.; Kienzle, B.; Zhao, D.; Feder, J.N. ‘Cold shock’ increases the frequency of homology directed repair gene editing in induced pluripotent stem cells. Sci. Rep. 2018, 8, 2080. [Google Scholar] [CrossRef]
- Canny, M.D.; Moatti, N.; Wan, L.C.K.; Fradet-Turcotte, A.; Krasner, D.; Mateos-Gomez, P.A.; Zimmermann, M.; Orthwein, A.; Juang, Y.C.; Zhang, W.; et al. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol. 2018, 36, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurihara, T.; Kouyama-Suzuki, E.; Satoga, M.; Li, X.; Badawi, M.; Thiha; Baig, D.N.; Yanagawa, T.; Uemura, T.; Mori, T.; et al. DNA repair protein RAD51 enhances the CRISPR/Cas9-mediated knock-in efficiency in brain neurons. Biochem. Biophys. Res. Commun. 2020, 524, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Molla, K.A.; Yang, Y. CRISPR/Cas-Mediated Base Editing: Technical Considerations and Practical Applications. Trends Biotechnol. 2019, 37, 1121–1142. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Désaulniers, K.; Ortiz, L.; Dufour, C.; Claudel, A.; Plourde, M.B.; Merindol, N.; Berthoux, L. Editing of the TRIM5 Gene Decreases the Permissiveness of Human T Lymphocytic Cells to HIV-1. Viruses 2021, 13, 24. https://doi.org/10.3390/v13010024
Désaulniers K, Ortiz L, Dufour C, Claudel A, Plourde MB, Merindol N, Berthoux L. Editing of the TRIM5 Gene Decreases the Permissiveness of Human T Lymphocytic Cells to HIV-1. Viruses. 2021; 13(1):24. https://doi.org/10.3390/v13010024
Chicago/Turabian StyleDésaulniers, Kevin, Levine Ortiz, Caroline Dufour, Alix Claudel, Mélodie B. Plourde, Natacha Merindol, and Lionel Berthoux. 2021. "Editing of the TRIM5 Gene Decreases the Permissiveness of Human T Lymphocytic Cells to HIV-1" Viruses 13, no. 1: 24. https://doi.org/10.3390/v13010024
APA StyleDésaulniers, K., Ortiz, L., Dufour, C., Claudel, A., Plourde, M. B., Merindol, N., & Berthoux, L. (2021). Editing of the TRIM5 Gene Decreases the Permissiveness of Human T Lymphocytic Cells to HIV-1. Viruses, 13(1), 24. https://doi.org/10.3390/v13010024