
Novel Fusari- and Toti-like Viruses, with Probable Different Origins, in the Plant Pathogenic Oomycete Globisporangium ultimum


Abstract
:1. Introduction
2. Materials and Methods
2.1. Globisporangium ultimum Isolates
2.2. dsRNA Extraction
2.3. High-Throughput Sequencing of dsRNA
2.4. Northern Blot Analysis for dsRNA
2.5. RT-PCR
2.6. Determination of dsRNA Genome
2.7. Bioinformatic Analysis
2.8. Phylogenetic Analyses
2.9. Codon Usage Bias Analysis
3. Results
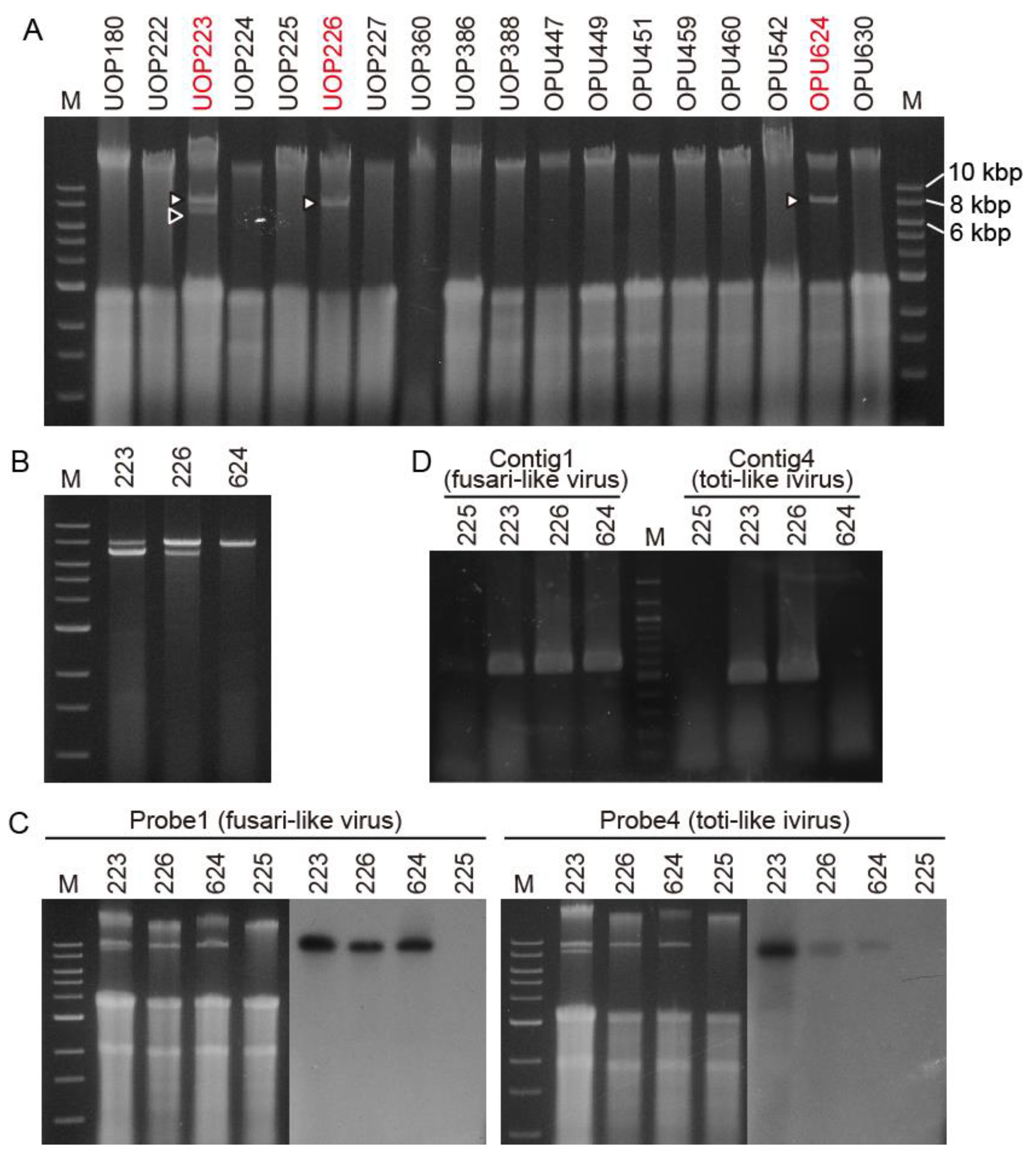
3.1. Detection of Viral-Like dsRNAs in the G. ultimum Isolates in Japan and Norway
3.2. Pythium ultimum RNA Virus 1 (PuRV1)
3.2.1. Genome Sequencing of PuRV1
3.2.2. Organization of PuRV1 Genome
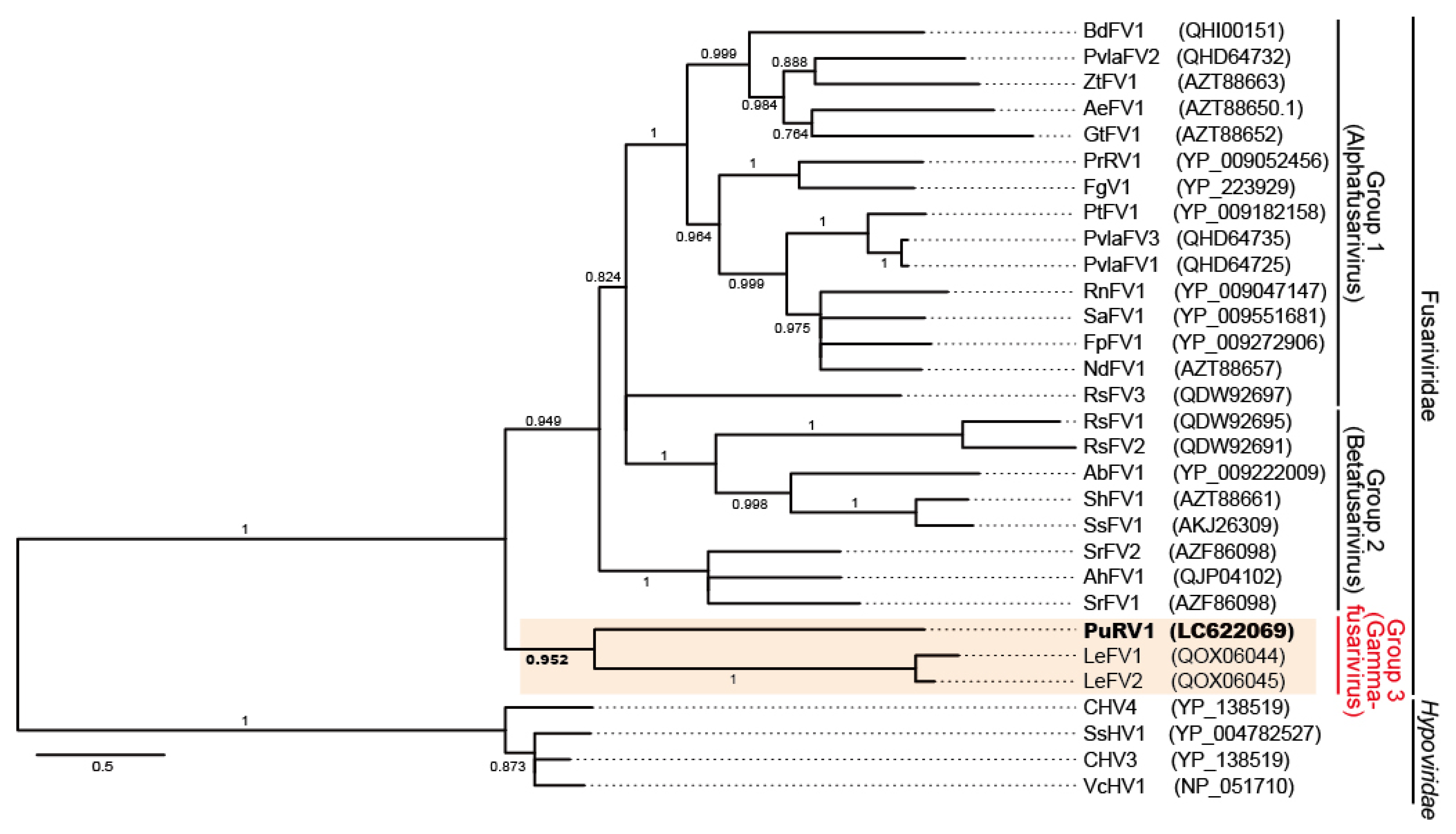
3.2.3. Phylogenetic Analysis of PuRV1
3.3. Pythium ultimum RNA Virus 2 (PuRV2)
3.3.1. Genome Sequencing of PuRV2
3.3.2. Organization of PuRV2 Genome
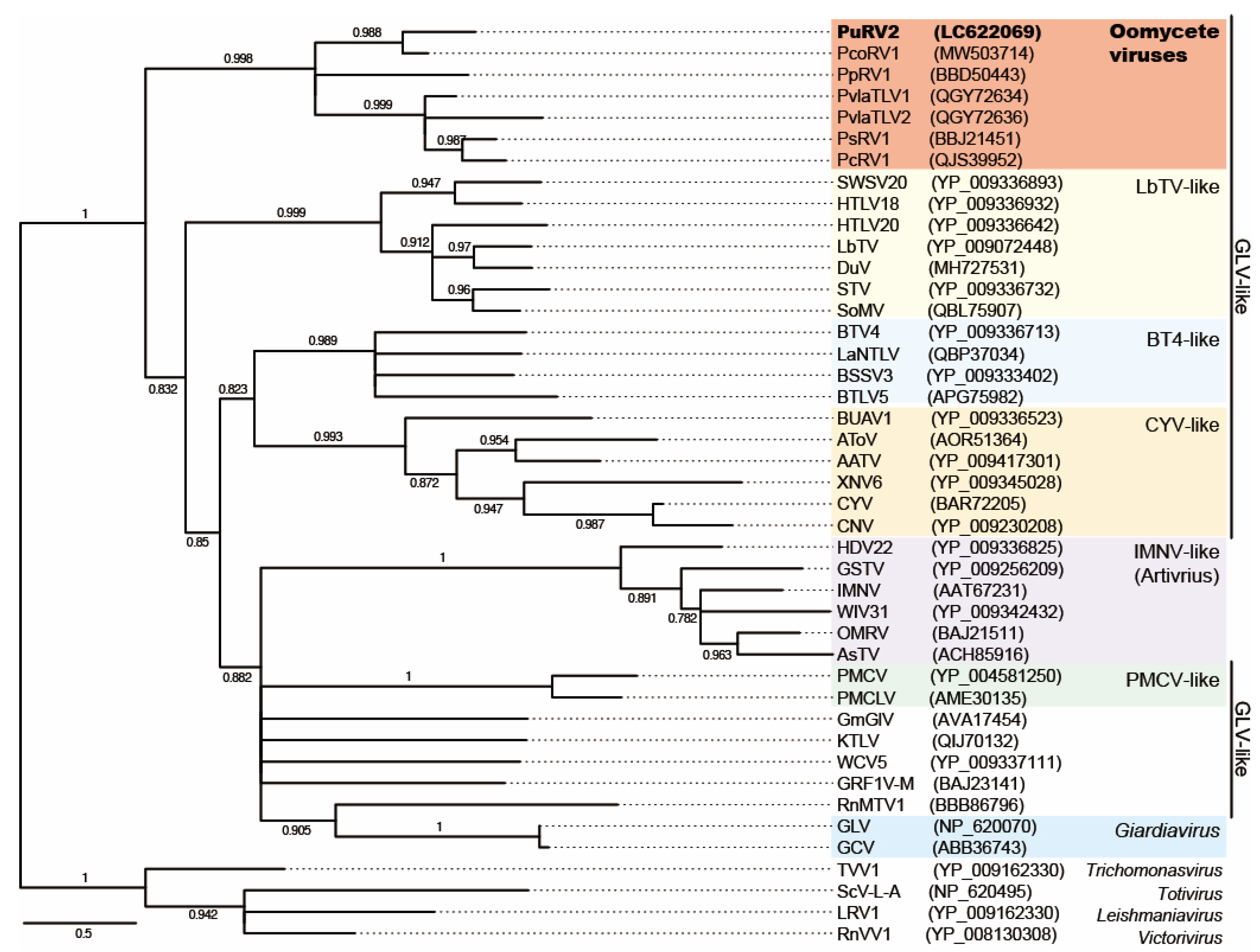
3.3.3. Phylogenetic Analysis of PuRV2
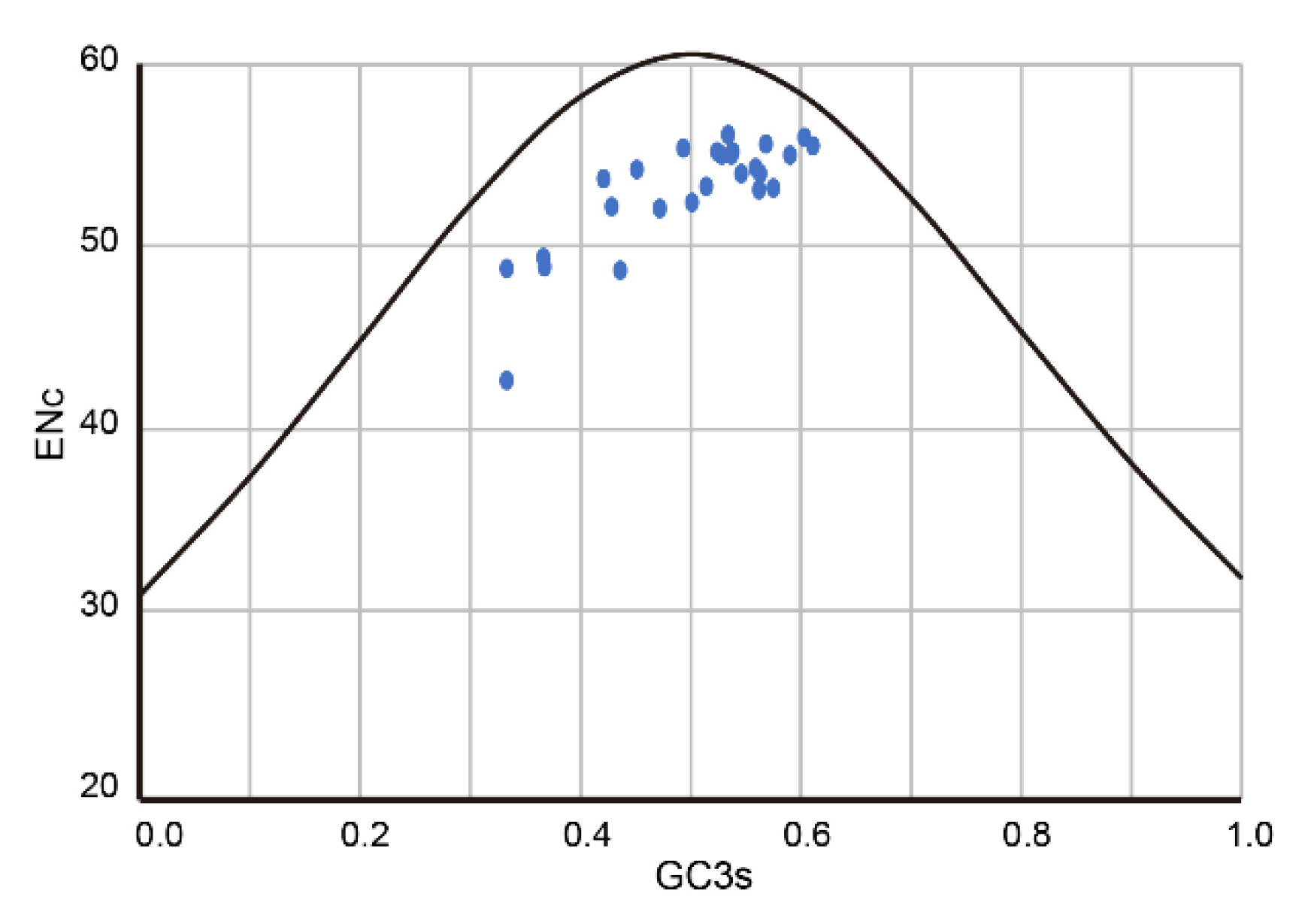
3.4. Codon Usage Bias Analysis of PuRV1
4. Discussion
4.1. Pythium ultimum RNA Virus 1 (PuRV1)
4.2. Pythium ultimum RNA Virus 2
4.3. Origins of PuRV1 and PuRV2
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamoun, S.; Furzer, O.; Jones, J.D.; Judelson, H.S.; Ali, G.S.; Dalio, R.J.; Roy, S.G.; Schena, L.; Zambounis, A.; Panabières, F.; et al. The Top 10 oomycete pathogens in molecular plant pathology. Mol. Plant. Pathol. 2015, 16, 413–434. [Google Scholar] [CrossRef] [PubMed]
- Thines, M.; Kamoun, S. Oomycete–plant coevolution: Recent advances and future prospects. Curr. Opin. Plant. Biol. 2010, 13, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Sutela, S.; Poimala, A.; Vainio, E.J. Viruses of fungi and oomycetes in the soil environment. FEMS Microbiol. Ecol. 2019, 95, fiz119. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Myers, K.; Hillman, B.I.; Fry, W.E. A novel virus of the late blight pathogen, Phytophthora infestans, with two RNA segments and a supergroup 1 RNA-dependent RNA polymerase. Virology 2009, 392, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Myers, K.; Fry, W.E.; Hillman, B.I. A member of the virus family Narnaviridae from the plant pathogenic oomycete Phytophthora infestans. Arch. Virol. 2012, 157, 165–169. [Google Scholar] [CrossRef]
- Cai, G.; Krychiw, J.F.; Myers, K.; Fry, W.E.; Hillman, B.I. A new virus from the plant pathogenic oomycete Phytophthora infestans with an 8 kb dsRNA genome: The sixth member of a proposed new virus genus. Virology 2013, 435, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Myers, K.; Fry, W.E.; Hillman, B.I. Phytophthora infestans RNA virus 2, a novel RNA virus from Phytophthora infestans, does not belong to any known virus group. Arch. Virol. 2019, 164, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Botella, L.; Jung, T. Multiple viral infections detected in Phytophthora condilina by total and small RNA sequencing. Viruses 2021, 13, 620. [Google Scholar] [CrossRef]
- Hacker, C.V.; Brasier, C.M.; Buck, K.W. A double-stranded RNA from a Phytophthora species is related to the plant endornaviruses and contains a putative UDP glycosyltransferase gene. J. Gen. Virol. 2005, 86, 1561–1570. [Google Scholar] [CrossRef]
- Kozlakidis, Z.; Brown, N.A.; Jamal, A.; Phoon, X.; Coutts, R.H.A. Incidence of endornaviruses in Phytophthora taxon douglasfir and Phytophthora ramorum. Virus Genes 2010, 40, 130–134. [Google Scholar] [CrossRef]
- Botella, L.; Janoušek, J.; Maia, C.; Jung, M.H.; Raco, M.; Jung, T. Marine oomycetes of the genus Halophytophthora harbor viruses related to Bunyaviruses. Front. Microbiol. 2020, 11, 1467. [Google Scholar] [CrossRef] [PubMed]
- Poimala, A.; Vainio, E.J. Complete genome sequence of a novel toti-like virus from the plant-pathogenic oomycete Phytophthora cactorum. Arch. Virol. 2020, 165, 1679–1682. [Google Scholar] [CrossRef] [PubMed]
- Poimala, A.; Parikka, P.; Hantula, J.; Vainio, E.J. Viral diversity in Phytophthora cactorum population infecting strawberry. Environ. Microbiol. 2021, 23. [Google Scholar] [CrossRef]
- Uchida, K.; Sakuta, K.; Ito, A.; Takahashi, Y.; Katayama, Y.; Omatsu, T.; Mizutani, T.; Arie, T.; Komatsu, K.; Fukuhara, T.; et al. Two novel endornaviruses co-infecting a Phytophthora pathogen of Asparagus officinalis modulate the developmental stages and fungicide sensitivities of the host oomycete. Front. Microbiol. 2021, 12, 633502. [Google Scholar] [CrossRef]
- Buck, K.W. Fungal virology-an overview. In Fungal Virology; Buck, K.W., Ed.; CRC Press: Boca Raton, FL, USA, 1986; pp. 1–84. [Google Scholar]
- Klassen, G.R.; Kim, W.K.; Barr, D.J.S.; Desaulniers, N.L. Presence of double-stranded RNA in isolates of Pythium irregulare. Mycologia 1991, 83, 657–661. [Google Scholar] [CrossRef]
- Gillings, M.R.; Tesoriero, L.A.; Gunn, L.V. Detection of double-stranded RNA and virus-like particles in Australian isolates of Pythium irregulare. Plant. Pathol. 1993, 42, 6–15. [Google Scholar] [CrossRef]
- Yokoi, T.; Takemoto, Y.; Suzuki, M.; Yamashita, S.; Hibi, T. The nucleotide sequence and genome organization of Sclerophthora macrospora virus B. Virology 1999, 264, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, T.; Yamashita, S.; Hibi, T. The nucleotide sequence and genome organization of Sclerophthora macrospora virus A. Virology 2003, 31, 394–399. [Google Scholar] [CrossRef] [Green Version]
- Heller-Dohmen, M.; Göpfert, J.C.; Pfannstiel, J.; Spring, O. The nucleotide sequence and genome organization of Plasmopara halstedii virus. Virol. J. 2011, 8, 123. [Google Scholar] [CrossRef] [Green Version]
- Chiapello, M.; Rodríguez-Romero, J.; Ayllón, M.A.; Turina, M. Analysis of the virome associated to grapevine downy mildew lesions reveals new mycovirus lineages. Virus Evol. 2020, 6, veaa058. [Google Scholar] [CrossRef]
- Shiba, K.; Hatta, C.; Sasai, S.; Tojo, M.; Ohki, S.T.; Mochizuki, T. Genome sequence of a novel partitivirus identified from the oomycete Pythium nunn. Arch. Virol. 2018, 163, 2561–2563. [Google Scholar] [CrossRef]
- Shiba, K.; Hatta, C.; Sasai, S.; Tojo, M.; Ohki, S.T.; Mochizuki, T. A novel toti-like virus from a plant pathogenic oomycete Globisporangium splendens. Virology 2019, 537, 165–171. [Google Scholar] [CrossRef]
- Sasai, S.; Tamura, K.; Tojo, M.; Herrero, M.L.; Hoshino, T.; Ohki, S.T.; Mochizuki, T. A novel non-segmented double-stranded RNA virus from an Arctic isolate of Pythium polare. Virology 2018, 522, 234–243. [Google Scholar] [CrossRef]
- Uzuhashi, S.; Tojo, M.; Kakishima, M. Phylogeny of the genus Pythium and description of new genera. Mycoscience 2010, 51, 337–365. [Google Scholar] [CrossRef]
- Domsch, K.H.; Gams, W.; Anderson, T.-H. Compendium of Soil Fungi; Academic Press: London, UK, 1980; Volume 1, pp. 1–857. [Google Scholar]
- Lerch-Olson, E.R.; Robertson, A.E. Effect of co-inoculations with Pythium and Fusarium species on seedling disease development of soybean. Can. J. Plant. Pathol. 2020, 42, 408–418. [Google Scholar] [CrossRef]
- Lévesque, C.A.; Brouwer, H.; Cano, L.; Hamilton, J.P.; Holt, C.; Huitema, E.; Robideau, G.P.; Thines, M.; Win, J.; Zerillo, M.M.; et al. Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol. 2010, 11, R73. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.J.; Dodds, J.A. Isolation and analysis of double-stranded RNA from virus-infected plant and fungal tissue. Phytopathology 1979, 69, 854–858. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 2019, 8, giz100. [Google Scholar] [CrossRef] [Green Version]
- Byun, Y.; Han, K. PseudoViewer3: Generating planar drawings of large-scale RNA structures with pseudoknots. Bioinformatics 2009, 25, 1435–1437. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gribskov, M. IRESpy: An XGBoost model for prediction of internal ribosome entry sites. BMC Bioinform. 2019, 20, 409. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Posada, D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- McInerney, J.O. GCUA: General Codon Usage Analysis. Bioinformatics 1998, 14, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.H.; Zhang, J.; Sun, D.J.; Ma, Q.; Chen, H.T.; Ma, L.N.; Ding, Y.Z.; Liu, Y.S. The distribution of synonymous codon choice in the translation initiation region of dengue virus. PLoS ONE 2013, 8, e77239. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index-a measure of directional synonymous codon usage bias, and its potential applications. Nucl. Acid Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Puigbò, P.; Aragonès, L.; Garcia-Vallvé, S. RCDI/eRCDI: A web-server to estimate codon usage deoptimization. BMC Res. Notes 2010, 3, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Shen, G.; Wang, J.; Liu, M.; Bian, Y.; Xu, Z. Mycoviral diversity and characteristics of a negative-stranded RNA virus LeNSRV1 in the edible mushroom Lentinula edodes. Virology 2021, 555, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Bruenn, J.A. A closely related group of RNA-dependent RNA polymerases from double-stranded RNA viruses. Nucl. Acid Res. 1993, 21, 5667–5669. [Google Scholar] [CrossRef] [PubMed]
- Bruenn, J.A. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucl. Acid Res. 2003, 31, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Salaipeth, L.; Lin, Y.H.; Sasaki, A.; Kanematsu, S.; Suzuki, N. A novel bipartite double-stranded RNA mycovirus from the white root rot fungus Rosellinia necatrix: Molecular and biological characterization, taxonomic considerations, and potential for biological control. J. Virol. 2009, 83, 12801–12812. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Fu, Y.; Xie, J.; Cheng, J.; Ghabrial, S.A.; Li, G.; Peng, Y.; Yi, X.; Jiang, D. Evolutionary genomics of mycovirus-related dsRNA viruses reveals cross-family horizontal gene transfer and evolution of diverse viral lineages. BMC Evol. Biol. 2012, 12, 91. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Shen, X.; Murphy, R.W.; Shen, Y. The adaptation of codon usage of + ssRNA viruses to their hosts. Infect. Genet. Evol. 2018, 63, 175–179. [Google Scholar] [CrossRef]
- Zhong, J.; Zhao, S.Q.; Li, G.F.; Pang, X.D.; Deng, X.J.; Zhu, H.J.; Da Gao, B.; Zhou, Q. A novel fusarivirus isolated from the phytopathogenic fungus Nigrospora oryzae. Virus Genes 2016, 52, 891–895. [Google Scholar] [CrossRef]
- Hrabáková, L.; Grum-Grzhimaylo, A.A.; Koloniuk, I.; Debets, A.J.M.; Sarkisova, T.; Petrzik, K. The alkalophilic fungus Sodiomyces alkalinus hosts beta- and gammapartitiviruses together with a new fusarivirus. PLoS ONE 2017, 12, e0187799. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, K.; Holcomb, E.E.; Allscheid, R.L.; Carrington, J. Hiding in plain sight: New virus genomes discovered via a systematic analysis of fungal public transcriptomes. PLoS ONE 2019, 14, e0219207. [Google Scholar] [CrossRef] [Green Version]
- Honda, S.; Eusebio-Cope, A.; Miyashita, S.; Yokoyama, A.; Aulia, A.; Shahi, S.; Kondo, H.; Suzuki, N. Establishment of Neurospora crassa as a model organism for fungal virology. Nat. Commun. 2020, 11, 5627. [Google Scholar] [CrossRef]
- Fuglsang, A. Impact of bias discrepancy and amino acid usage on estimates of the effective number of codons used in a gene, and a test for selection on codon usage. Gene 2008, 410, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.J.; Lim, W.S.; Park, S.H.; Park, M.R.; Kim, K.H. Molecular characterization of a dsRNA mycovirus, Fusarium graminearum virus-DK21, which is phylogenetically related to hypoviruses but has a genome organization and gene expression strategy resembling those of plant potex-like viruses. Mol. Cells 2007, 23, 304–315. [Google Scholar] [CrossRef]
- De Lima, J.G.S.; Teixeira, D.G.; Freitas, T.T.; Lima, J.P.M.S.; Lanza, D.C.F.R. Evolutionary origin of 2A-like sequences in Totiviridae genomes. Virus Res. 2019, 259, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Charon, J.; Murray, S.; Holmes, E.C. Revealing RNA virus diversity and evolution in unicellular algae transcriptomes. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ghabrial, S.A. Origin, adaptation and evolutionary pathways of fungal viruses. Virus Genes 1998, 16, 119–131. [Google Scholar] [CrossRef]
- Naitow, H.; Tang, J.; Canady, M.; Wickner, R.B.; Johnson, J.E. L-A virus at 3.4 Å resolution reveals particle architecture and mRNA decapping mechanism. Nat. Struct. Biol. 2002, 9, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Spear, A.; Sisterson, M.S.; Yokomi, R.; Stenger, D.C. Plant-feeding insects harbor double-stranded RNA viruses encoding a novel proline-alanine rich protein and a polymerase distantly related to that of fungal viruses. Virology 2010, 404, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, T.; Esteban, R. Cap-snatching mechanism in yeast L-A double-stranded RNA virus. Proc. Natl. Acad. Sci. USA 2011, 108, 17667–17671. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, D.J.; DeRosa, K.; Duffy, S. Base composition and translational selection are insufficient to explain codon usage bias in plant viruses. Viruses 2013, 5, 162–181. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shaul, A.; Gelbart, W.M. Viral ssRNAs are indeed compact. Biophys. J. 2015, 108, 14–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubiana, L.; Božič, A.L.; Micheletti, C.; Podgornik, R. Synonymous mutations reduce genome compactness in icosahedral ssRNA viruses. Biophys. J. 2015, 108, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, T.; Ohara, R.; Roossinck, M.J. Large-scale synonymous substitutions in cucumber mosaic virus RNA 3 facilitate amino acid mutations in the coat protein. J. Virol. 2018, 92, e01007-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roossinck, M.J. Evolutionary and ecological links between plant and fungal viruses. New Phytol. 2019, 221, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Hassani, M.A.; Durán, P.; Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef]
- Jiang, R.H.; Tyler, B.M. Mechanisms and evolution of virulence in oomycetes. Annu. Rev. Phytopathol. 2012, 50, 295–318. [Google Scholar] [CrossRef]
- Ma, X.; Li, H.; O’Rourke, T.; Sivasithamparam, K.; Barbetti, M.J. Co-occurrence of an Aphanomyces sp. and Phytopththora clandestina in subterranean clover pastures in the high rainfall areas of the lower south-west of Western Australia. Australas. Plant. Pathol. 2008, 37, 74–78. [Google Scholar] [CrossRef]
- Lo Giudice, V.; Raudino, F.; Magnano di San Lio, R.; Cacciola, S.O.; Faedda, R.; Pane, A. First report of a decline and wilt of young olive trees caused by simultaneous infections of Verticillium dahliae and Phytophthora palmivora in Sicily. Plant. Dis. 2010, 94, 1372. [Google Scholar] [CrossRef] [PubMed]
- Weiland, J.E.; Benedict, C.; Zasada, I.A.; Scagel, C.R.; Beck, B.R.; Davis, A.; Graham, K.; Peetz, A.; Martin, R.R.; Dung, J.K.S.; et al. Late-summer disease symptoms in western washington red raspberry fields associated with co-occurrence of Phytophthora rubi, Verticillium dahliae, and Pratylenchus penetrans, but not Raspberry bushy dwarf virus. Plant. Dis. 2018, 102, 938–947. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Query Cover | E-Value | Per. Ident | Accession |
|---|---|---|---|---|
| RNA-dependent RNA polymerase (Phytophthora condilina RNA virus 1) | 92% | 0.0 | 60.6 | QTT60989.1 |
| RNA-dependent RNA polymerase (Pythium polare RNA virus 1) | 88% | 9.00 × 10−87 | 34.9 | YP_009552275.1 |
| Putative RNA-dependent RNA polymerase (Plasmopara viticola lesion-associated totivirus-like 1) | 63% | 1.00 × 10−67 | 34.1 | QGY72634.1 |
| Putative RNA-dependent RNA polymerase (Plasmopara viticola lesion-associated totivirus-like 2) | 58% | 1.00 × 10−64 | 34.9 | QGY72636.1 |
| CP-RdRp fusion protein (Phytophthora cactorum RNA virus 1) | 62% | 5.00 × 10−61 | 33.4 | QJS39952.1 |
| CP-RdRp fusion protein (Pythium splendens RNA virus 1) | 63% | 8.00 × 10−55 | 32.8 | BBJ21453.1 |
| CP-RdRp fusion protein (Pythium splendens RNA virus 1) | 63% | 1.00 × 10−54 | 32.8 | BBJ21451.1 |
| PuRV2 | PpRV1 | PsRV1 | PcRV1 | PcoRV1 | PvlaTLV1 | PvlaTLV2 | |
|---|---|---|---|---|---|---|---|
| PuRV2 | 41.5 | 35.2 | 35.3 | 63.9 | 36.8 | 35.4 | |
| PpRV1 | no a | 37.8 | 38.4 | 41.2 | 39.0 | 33.9 | |
| PsRV1 | no | no | 65.9 | 36.2 | 59.9 | 48.3 | |
| PcRV1 | no | no | 68.0 | 35.5 | 57.0 | 49.0 | |
| PcoRV1 | 65.0 | no | no | no | 38.8 | 38.1 | |
| PvlaTLV1 | no | no | no | no | no | 49.4 | |
| PvlaTLV2 | no | no | no | no | no | no |
| Virus a | Length (nt) | Codon Adaptation Index (CAI) | GC% | ENc b | ||||
|---|---|---|---|---|---|---|---|---|
| Globisporangium ultimum | Phytophthora cactorum | Plasmopara viticola | Saccharomyces cerevisiae | Lentinula edodes | ||||
| PuRV1 | 6303 | 0.522 | 0.657 | 0.816 | 0.761 | 0.855 | 42.9 | 48.7 |
| LeFV1 | 5616 | 0.455 | 0.614 | 0.826 | 0.813 | 0.874 | 39.6 | 42.7 |
| LeFV2 | 3591 | 0.469 | 0.623 | 0.802 | 0.786 | 0.866 | 41.5 | 48.9 |
| PvlaFV1 | 4650 | 0.528 | 0.664 | 0.772 | 0.727 | 0.848 | 46.0 | 53.3 |
| PvlaFV2 | 4614 | 0.550 | 0.687 | 0.756 | 0.697 | 0.843 | 50.8 | 54.0 |
| PvlaFV3 | 4509 | 0.508 | 0.646 | 0.776 | 0.742 | 0.869 | 45.2 | 52.1 |
| AeFV1 | 4671 | 0.575 | 0.707 | 0.750 | 0.678 | 0.823 | 52.2 | 55.5 |
| SaFV1 | 4584 | 0.534 | 0.671 | 0.754 | 0.707 | 0.832 | 49.2 | 54.0 |
| NdFv1 | 4581 | 0.554 | 0.686 | 0.761 | 0.705 | 0.842 | 49.0 | 55.6 |
| FpFV1 | 4506 | 0.553 | 0.692 | 0.764 | 0.707 | 0.842 | 49.1 | 54.3 |
| RnFV1 | 4629 | 0.560 | 0.691 | 0.788 | 0.722 | 0.851 | 46.6 | 55.0 |
| PtFV1 | 4647 | 0.532 | 0.668 | 0.789 | 0.738 | 0.852 | 45.3 | 55.4 |
| ZtFV1 | 4476 | 0.573 | 0.698 | 0.758 | 0.688 | 0.835 | 51.0 | 55.0 |
| GtFV1 | 4578 | 0.542 | 0.683 | 0.771 | 0.710 | 0.848 | 49.1 | 55.0 |
| BdFV1 | 4635 | 0.542 | 0.674 | 0.792 | 0.740 | 0.860 | 46.3 | 52.4 |
| PrRV1 | 4572 | 0.558 | 0.684 | 0.768 | 0.697 | 0.851 | 49.5 | 53.1 |
| FgV1 | 4653 | 0.571 | 0.705 | 0.760 | 0.696 | 0.848 | 51.4 | 53.2 |
| SrFV1 | 4914 | 0.513 | 0.657 | 0.797 | 0.742 | 0.856 | 45.4 | 54.2 |
| AhFV1 | 4914 | 0.578 | 0.702 | 0.741 | 0.668 | 0.823 | 52.3 | 56.0 |
| SrFV2 | 5037 | 0.539 | 0.675 | 0.760 | 0.699 | 0.836 | 49.0 | 55.2 |
| RsFV3 | 5388 | 0.537 | 0.675 | 0.766 | 0.711 | 0.849 | 47.6 | 55.2 |
| AbFV1 | 4569 | 0.555 | 0.682 | 0.773 | 0.713 | 0.841 | 47.1 | 56.1 |
| ShFV1 | 5157 | 0.446 | 0.583 | 0.798 | 0.801 | 0.853 | 36.3 | 48.8 |
| SsFV1 | 4998 | 0.472 | 0.604 | 0.812 | 0.787 | 0.856 | 37.7 | 49.4 |
| RsFV1 | 4683 | 0.499 | 0.632 | 0.796 | 0.757 | 0.850 | 41.9 | 52.2 |
| RsFV2 | 4587 | 0.493 | 0.625 | 0.793 | 0.764 | 0.853 | 40.7 | 53.7 |
| Virus a | Relative Codon Deoptimization Index (RCDI) | ||||
|---|---|---|---|---|---|
| Globisporangium ultimum | Phytophthora cactorum | Plasmopara viticola | Saccharomyces cerevisiae | Lentinula edodes | |
| PuRV1 | 1.935 | 1.549 | 1.269 | 1.127 | 1.233 |
| LeFV1 | 2.520 | 1.852 | 1.316 | 1.205 | 1.333 |
| LeFV2 | 2.222 | 1.655 | 1.252 | 1.140 | 1.204 |
| PvlaFV1 | 1.809 | 1.395 | 1.325 | 1.172 | 1.148 |
| PvlaFV2 | 1.521 | 1.252 | 1.282 | 1.332 | 1.139 |
| PvlaFV3 | 1.888 | 1.461 | 1.316 | 1.220 | 1.133 |
| PtFV1 | 1.567 | 1.273 | 1.143 | 1.143 | 1.089 |
| ZtFV1 | 1.361 | 1.188 | 1.255 | 1.330 | 1.147 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukunishi, M.; Sasai, S.; Tojo, M.; Mochizuki, T. Novel Fusari- and Toti-like Viruses, with Probable Different Origins, in the Plant Pathogenic Oomycete Globisporangium ultimum. Viruses 2021, 13, 1931. https://doi.org/10.3390/v13101931
Fukunishi M, Sasai S, Tojo M, Mochizuki T. Novel Fusari- and Toti-like Viruses, with Probable Different Origins, in the Plant Pathogenic Oomycete Globisporangium ultimum. Viruses. 2021; 13(10):1931. https://doi.org/10.3390/v13101931
Chicago/Turabian StyleFukunishi, Miki, Shinsaku Sasai, Motoaki Tojo, and Tomofumi Mochizuki. 2021. "Novel Fusari- and Toti-like Viruses, with Probable Different Origins, in the Plant Pathogenic Oomycete Globisporangium ultimum" Viruses 13, no. 10: 1931. https://doi.org/10.3390/v13101931
APA StyleFukunishi, M., Sasai, S., Tojo, M., & Mochizuki, T. (2021). Novel Fusari- and Toti-like Viruses, with Probable Different Origins, in the Plant Pathogenic Oomycete Globisporangium ultimum. Viruses, 13(10), 1931. https://doi.org/10.3390/v13101931