
Precursors of Viral Proteases as Distinct Drug Targets


Abstract
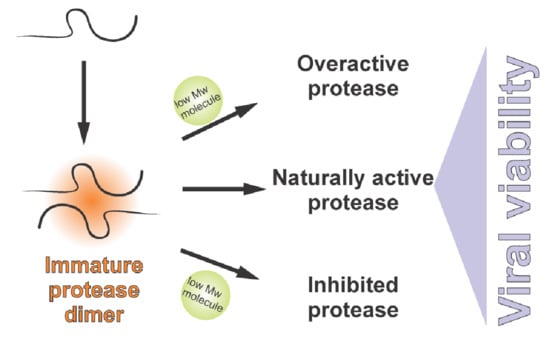
:
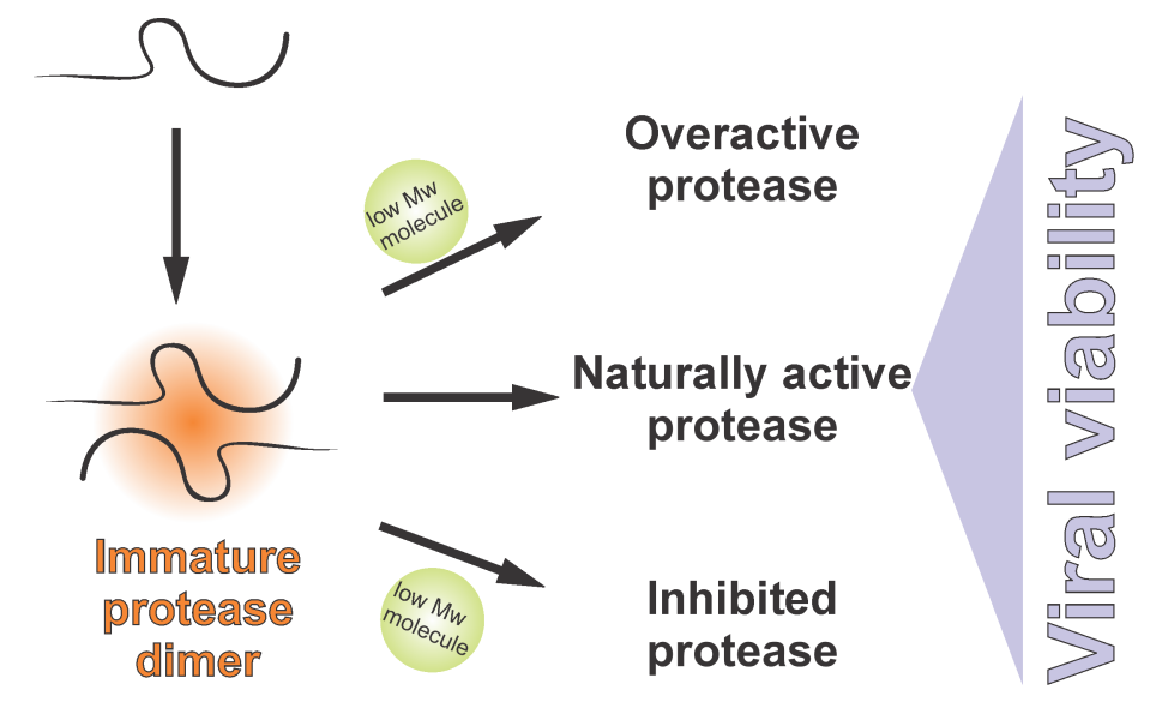{kind=link}
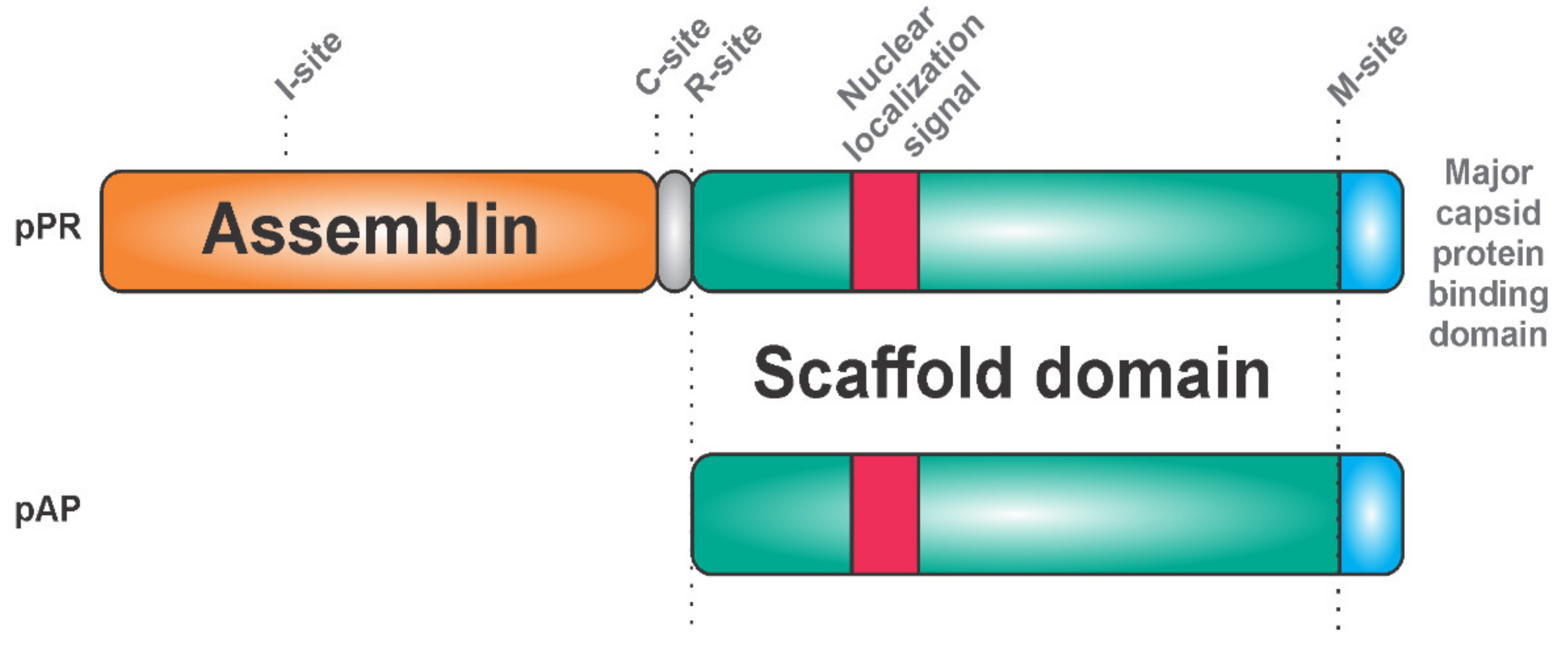{kind=link}
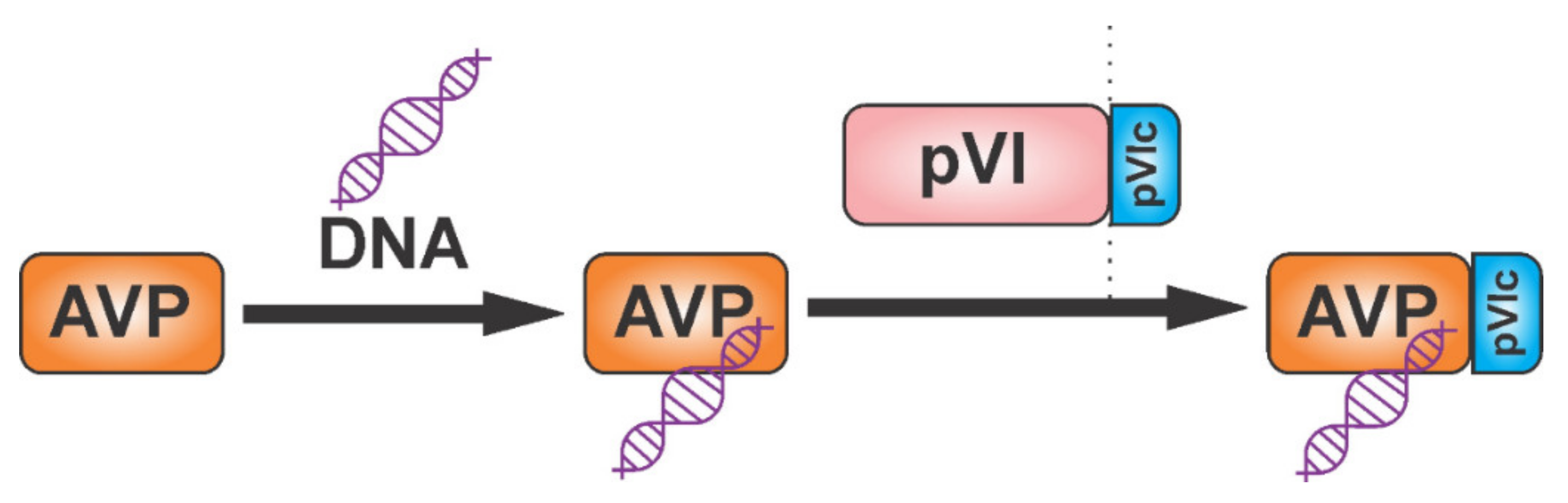{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Precursors as Major Signalization and Orchestration Agents
Viral Polyprotein Strategy
2. DNA Viruses
2.1. Herpesviruses
2.2. Adenoviruses
3. RNA Viruses
3.1. Retroviruses
3.2. Picornaviruses
3.3. Caliciviruses
3.4. Togaviruses
3.5. Flaviviruses
3.6. Coronaviruses
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kassell, B.; Kay, J. Zymogens of Proteolytic Enzymes. Science 1973, 180, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Keil, B.; Meloun, B.; Vanecek, J.; Kostka, V.; Prusik, Z.; Sorm, F. Partial structure of chymotrypsinogen. Biochim. Biophys. Acta 1962, 56, 595–599. [Google Scholar] [CrossRef]
- Meloun, B.; Kluh, I.; Kostka, V.; Morávek, L.; Prusík, Z.; Vanĕcek, J.; Keil, B.; Sorm, F. Covalent structure of bovine chymotrypsinogen A. Biochim. Biophys. Acta 1966, 130, 543–546. [Google Scholar] [CrossRef]
- Keilova, H.; Kostka, V.; Kay, J. The first step in the activation of chicken pepsinogen is similar to that of prochymosin. Biochem. J. 1977, 167, 855–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichová, I.; Kostka, V. Molecular characteristics of pepsinogen and pepsin from duck glandular stomach. Comp. Biochem. Physiol. B 1990, 97, 89–94. [Google Scholar] [CrossRef]
- Strop, P.; Cechová, D.; Tomásek, V. Model study of hydrophobic interactions of alpha- and beta-trypsin and alpha-chymotrypsin. J. Chromatogr. 1983, 259, 255–268. [Google Scholar] [CrossRef]
- Hynek, R.; Kasicka, V.; Kucerová, Z.; Kás, J. Fast detection of phosphorylation of human pepsinogen A, human pepsinogen C and swine pepsinogen using a combination of reversed-phase high-performance liquid chromatography and capillary zone electrophoresis for peptide mapping. J. Chromatogr. B Biomed. Sci. Appl. 1997, 688, 213–220. [Google Scholar] [CrossRef]
- Gómez-Outes, A.; Suárez-Gea, M.L.; Calvo-Rojas, G.; Lecumberri, R.; Rocha, E.; Pozo-Hernández, C.; Terleira-Fernández, A.I.; Vargas-Castrillón, E. Discovery of anticoagulant drugs: A historical perspective. Curr. Drug Discov. Technol. 2012, 9, 83–104. [Google Scholar] [CrossRef]
- Foltmann, B. A review on prorennin and rennin. C. R. Trav. Lab. Carlsberg 1966, 35, 143–231. [Google Scholar]
- Hasilik, A.; von Figura, K.; Conzelmann, E.; Nehrkorn, H.; Sandhoff, K. Lysosomal enzyme precursors in human fibroblasts. Activation of cathepsin D precursor in vitro and activity of beta-hexosaminidase A precursor towards ganglioside GM2. Eur. J. Biochem. 1982, 125, 317–321. [Google Scholar] [CrossRef]
- Mása, M.; Maresová, L.; Vondrásek, J.; Horn, M.; Jezek, J.; Mares, M. Cathepsin D propeptide: Mechanism and regulation of its interaction with the catalytic core. Biochemistry 2006, 45, 15474–15482. [Google Scholar] [CrossRef]
- Hánová, I.; Brynda, J.; Houštecká, R.; Alam, N.; Sojka, D.; Kopáček, P.; Marešová, L.; Vondrášek, J.; Horn, M.; Schueler-Furman, O.; et al. Novel Structural Mechanism of Allosteric Regulation of Aspartic Peptidases via an Evolutionarily Conserved Exosite. Cell Chem. Biol. 2018, 25, 318–329.e314. [Google Scholar] [CrossRef] [Green Version]
- Houštecká, R.; Hadzima, M.; Fanfrlík, J.; Brynda, J.; Pallová, L.; Hánová, I.; Mertlíková-Kaiserová, H.; Lepšík, M.; Horn, M.; Smrčina, M.; et al. Biomimetic Macrocyclic Inhibitors of Human Cathepsin D: Structure-Activity Relationship and Binding Mode Analysis. J. Med. Chem. 2020, 63, 1576–1596. [Google Scholar] [CrossRef]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol Modification of Hedgehog Signaling Proteins in Animal Development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef]
- Shinde, U.; Fu, X.; Inouye, M. A Pathway for Conformational Diversity in Proteins Mediated by Intramolecular Chaperones. J. Biol. Chem. 1999, 274, 15615–15621. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.S.; Banerjee, S.; Aponte-Diaz, D.; Sharma, S.D.; Aligo, J.; Lodeiro, M.F.; Ning, G.; Sharma, R.; Arnold, J.J.; Cameron, C.E. Multiple poliovirus-induced organelles suggested by comparison of spatiotemporal dynamics of membranous structures and phosphoinositides. PLoS Pathg. 2018, 14, e1007036. [Google Scholar] [CrossRef] [Green Version]
- Konvalinka, J.; Kräusslich, H.G.; Müller, B. Retroviral proteases and their roles in virion maturation. Virology 2015, 479–480, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Renner, M.; Dejnirattisai, W.; Carrique, L.; Martin, I.S.; Karia, D.; Ilca, S.L.; Ho, S.F.; Kotecha, A.; Keown, J.R.; Mongkolsapaya, J.; et al. Flavivirus maturation leads to the formation of an occupied lipid pocket in the surface glycoproteins. Nat. Commun. 2021, 12, 1238. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Monastyrska, I.; Griffith, J.; van der Sluijs, P.; Voortman, J.; van Bergen en Henegouwen, P.M.; Vonk, A.M.; Rottier, P.J.; Reggiori, F.; de Haan, C.A. Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology 2014, 458–459, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Aleshin, A.E.; Drag, M.; Gombosuren, N.; Wei, G.; Mikolajczyk, J.; Satterthwait, A.C.; Strongin, A.Y.; Liddington, R.C.; Salvesen, G.S. Activity, Specificity, and Probe Design for the Smallpox Virus Protease K7L. J. Biol. Chem. 2012, 287, 39470–39479. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Culp, J.S.; DiLella, A.G.; Hellmig, B.; Hoog, S.S.; Janson, C.A.; Smith, W.W.; Abdel-Meguid, S.S. Unique fold and active site in cytomegalovirus protease. Nature 1996, 383, 275–279. [Google Scholar] [CrossRef]
- Saribas, A.S.; Coric, P.; Bouaziz, S.; Safak, M. Expression of novel proteins by polyomaviruses and recent advances in the structural and functional features of agnoprotein of JC virus, BK virus, and simian virus 40. J. Cell Physiol. 2019, 234, 8295–8315. [Google Scholar] [CrossRef]
- Hejtmánková, A.; Roubalová, K.; Forejtová, A.; Žáčková Suchanová, J.; Forstová, J.; Viklický, O.; Španielová, H. Prevalence of antibodies against BKPyV subtype I and IV in kidney transplant recipients and in the general Czech population. J. Med. Virol. 2019, 91, 856–864. [Google Scholar] [CrossRef]
- Majerová, T.; Hoffman, H.; Majer, F. Therapeutic targets for influenza-Perspectives in drug development. Collect. Czechoslov. Chem. Commun. 2010, 75, 81–103. [Google Scholar] [CrossRef]
- Yip, W.K.W.; Cristi, F.; Trifonov, G.; Narayan, N.; Kubanski, M.; Shmulevitz, M. The reovirus μ2 C-terminal loop inversely regulates NTPase and transcription functions versus binding to factory-forming μNS and promotes replication in tumorigenic cells. J. Virol. 2021, 95, e02006-20. [Google Scholar] [CrossRef]
- Zając, M.; Muszalska, I.; Sobczak, A.; Dadej, A.; Tomczak, S.; Jelińska, A. Hepatitis C—New drugs and treatment prospects. Eur. J. Med. Chem. 2019, 165, 225–249. [Google Scholar] [CrossRef]
- Gable, J.E.; Acker, T.M.; Craik, C.S. Current and Potential Treatments for Ubiquitous but Neglected Herpesvirus Infections. Chem. Rev. 2014, 114, 11382–11412. [Google Scholar] [CrossRef] [Green Version]
- Clercq, E.D.; Sakuma, T.; Baba, M.; Pauwels, R.; Balzarini, J.; Rosenberg, I.; Holý, A. Antiviral activity of phosphonylmethoxyalkyl derivatives of purine and pyrimidines. Antivir. Res. 1987, 8, 261–272. [Google Scholar] [CrossRef]
- Snoeck, R.; Sakuma, T.; De Clercq, E.; Rosenberg, I.; Holy, A. (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, a potent and selective inhibitor of human cytomegalovirus replication. Antimicrob. Agents Chemother. 1988, 32, 1839–1844. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Tsuge, H.; Almassy, R.J.; Gribskov, C.L.; Katoh, S.; Vanderpool, D.L.; Margosiak, S.A.; Pinko, C.; Matthews, D.A.; Kan, C.-C. Structure of the Human Cytomegalovirus Protease Catalytic Domain Reveals a Novel Serine Protease Fold and Catalytic Triad. Cell 1996, 86, 835–843. [Google Scholar] [CrossRef] [Green Version]
- Darke, P.L.; Cole, J.L.; Waxman, L.; Hall, D.L.; Sardana, M.K.; Kuo, L.C. Active human cytomegalovirus protease is a dimer. J. Biol. Chem. 1996, 271, 7445–7449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, L.; Qian, C.; Massariol, M.J.; Bonneau, P.R.; Cordingley, M.G.; Lagacé, L. A new serine-protease fold revealed by the crystal structure of human cytomegalovirus protease. Nature 1996, 383, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Shieh, H.S.; Kurumbail, R.G.; Stevens, A.M.; Stegeman, R.A.; Sturman, E.J.; Pak, J.Y.; Wittwer, A.J.; Palmier, M.O.; Wiegand, R.C.; Holwerda, B.C.; et al. Three-dimensional structure of human cytomegalovirus protease. Nature 1996, 383, 279–282. [Google Scholar] [CrossRef]
- Liu, F.Y.; Roizman, B. The herpes simplex virus 1 gene encoding a protease also contains within its coding domain the gene encoding the more abundant substrate. J. Virol. 1991, 65, 5149–5156. [Google Scholar] [CrossRef] [Green Version]
- Oien, N.L.; Thomsen, D.R.; Wathen, M.W.; Newcomb, W.W.; Brown, J.C.; Homa, F.L. Assembly of herpes simplex virus capsids using the human cytomegalovirus scaffold protein: Critical role of the C terminus. J. Virol. 1997, 71, 1281–1291. [Google Scholar] [CrossRef] [Green Version]
- Sheaffer, A.K.; Newcomb, W.W.; Brown, J.C.; Gao, M.; Weller, S.K.; Tenney, D.J. Evidence for controlled incorporation of herpes simplex virus type 1 UL26 protease into capsids. J. Virol. 2000, 74, 6838–6848. [Google Scholar] [CrossRef] [Green Version]
- Baum, E.Z.; Bebernitz, G.A.; Hulmes, J.D.; Muzithras, V.P.; Jones, T.R.; Gluzman, Y. Expression and analysis of the human cytomegalovirus UL80-encoded protease: Identification of autoproteolytic sites. J. Virol. 1993, 67, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Holwerda, B.C.; Wittwer, A.J.; Duffin, K.L.; Smith, C.; Toth, M.V.; Carr, L.S.; Wiegand, R.C.; Bryant, M.L. Activity of two-chain recombinant human cytomegalovirus protease. J. Biol. Chem. 1994, 269, 25911–25915. [Google Scholar] [CrossRef]
- Loveland, A.N.; Chan, C.K.; Brignole, E.J.; Gibson, W. Cleavage of human cytomegalovirus protease pUL80a at internal and cryptic sites is not essential but enhances infectivity. J. Virol. 2005, 79, 12961–12968. [Google Scholar] [CrossRef] [Green Version]
- Shimba, N.; Nomura, A.M.; Marnett, A.B.; Craik, C.S. Herpesvirus protease inhibition by dimer disruption. J. Virol. 2004, 78, 6657–6665. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.M.; Shahian, T.; Baharuddin, A.; Gable, J.E.; Craik, C.S. Enzyme Inhibition by Allosteric Capture of an Inactive Conformation. J. Mol. Biol. 2011, 411, 999–1016. [Google Scholar] [CrossRef] [Green Version]
- Shahian, T.; Lee, G.M.; Lazic, A.; Arnold, L.A.; Velusamy, P.; Roels, C.M.; Guy, R.K.; Craik, C.S. Inhibition of a viral enzyme by a small-molecule dimer disruptor. Nat. Chem. Biol. 2009, 5, 640–646. [Google Scholar] [CrossRef]
- Yamanaka, G.; DiIanni, C.L.; O’Boyle, D.R., II; Stevens, J.; Weinheimer, S.P.; Deckman, I.C.; Matusick-Kumar, L.; Colonno, R.J. Stimulation of the Herpes Simplex Virus Type I Protease by Antichaeotrophic Salts. J. Biol. Chem. 1995, 270, 30168–30172. [Google Scholar] [CrossRef] [Green Version]
- Kattenhorn, L.M.; Korbel, G.A.; Kessler, B.M.; Spooner, E.; Ploegh, H.L. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 2005, 19, 547–557. [Google Scholar] [CrossRef]
- Wang, J.; Loveland, A.N.; Kattenhorn, L.M.; Ploegh, H.L.; Gibson, W. High-molecular-weight protein (pUL48) of human cytomegalovirus is a competent deubiquitinating protease: Mutant viruses altered in its active-site cysteine or histidine are viable. J. Virol. 2006, 80, 6003–6012. [Google Scholar] [CrossRef] [Green Version]
- Schlieker, C.; Korbel, G.A.; Kattenhorn, L.M.; Ploegh, H.L. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J. Virol. 2005, 79, 15582–15585. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.T.; Oh, S.E.; Lee, Y.O.; Gibson, W.; Ahn, J.H. Cleavage specificity of the UL48 deubiquitinating protease activity of human cytomegalovirus and the growth of an active-site mutant virus in cultured cells. J. Virol. 2009, 83, 12046–12056. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Abrogates NF-κB Activation in DNA Sensing Signal Pathway. J. Virol. 2017, 91, e02417-16. [Google Scholar] [CrossRef] [Green Version]
- Westergren Jakobsson, A.; Segerman, B.; Wallerman, O.; Bergström Lind, S.; Zhao, H.; Rubin, C.-J.; Pettersson, U.; Akusjärvi, G. The Human Adenovirus 2 Transcriptome: An Amazing Complexity of Alternatively Spliced mRNAs. J. Virol. 2021, 95, e01869-20. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.P.; Mathews, M.B. DNA replication and the early to late transition in adenovirus infection. Cell 1980, 22, 523–533. [Google Scholar] [CrossRef]
- Donovan-Banfield, I.a.; Turnell, A.S.; Hiscox, J.A.; Leppard, K.N.; Matthews, D.A. Deep splicing plasticity of the human adenovirus type 5 transcriptome drives virus evolution. Commun. Biol. 2020, 3, 124. [Google Scholar] [CrossRef]
- Pied, N.; Wodrich, H. Imaging the adenovirus infection cycle. FEBS Lett. 2019, 593, 3419–3448. [Google Scholar] [CrossRef] [Green Version]
- Webster, A.; Hay, R.T.; Kemp, G. The adenovirus protease is activated by a virus-coded disulphide-linked peptide. Cell 1993, 72, 97–104. [Google Scholar] [CrossRef]
- Mangel, W.F.; McGrath, W.J.; Toledo, D.L.; Anderson, C.W. Viral DNA and a viral peptide can act as cofactors of adenovirus virion proteinase activity. Nature 1993, 361, 274–275. [Google Scholar] [CrossRef]
- Baniecki, M.L.; McGrath, W.J.; Mangel, W.F. Regulation of a Viral Proteinase by a Peptide and DNA in One-dimensional Space: III. Atomic resolution structure of the nascent form of the adenoirus proteinase. J. Biol. Chem. 2013, 288, 2081–2091. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.T.; McBride, K.M.; Baniecki, M.L.; Reich, N.C.; Marriott, G.; Mangel, W.F. Actin can act as a cofactor for a viral proteinase in the cleavage of the cytoskeleton. J. Biol. Chem. 2002, 277, 46298–46303. [Google Scholar] [CrossRef] [Green Version]
- Ruzindana-Umunyana, A.; Sircar, S.; Weber, J.M. The Effect of Mutant Peptide Cofactors on Adenovirus Protease Activity and Virus Infection. Virology 2000, 270, 173–179. [Google Scholar] [CrossRef]
- Barré-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [Green Version]
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Voisset, C.; Weiss, R.A.; Griffiths, D.J. Human RNA Viruses: The Search for Novel Human Retroviruses in Chronic Disease. Microbiol Mol. Biol. Rev. 2008, 72, 157–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Clercq, E. HIV resistance to reverse transcriptase inhibitors. Biochem. Pharmacol. 1994, 47, 155–169. [Google Scholar] [CrossRef]
- Holý, A.; Rosenberg, I. Synthesis of 9-(2-phosphonylmethoxyethyl)adenine and related compounds. Collect. Czechoslov. Chem. Commun. 1987, 52, 2801–2809. [Google Scholar] [CrossRef]
- Balzarini, J.; Holy, A.; Jindrich, J.; Naesens, L.; Snoeck, R.; Schols, D.; De Clercq, E. Differential antiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates: Potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethoxypropyl)-2,6-diaminopurine. Antimicrob. Agents Chemother. 1993, 37, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A.V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Kröhn, A.; et al. Rational design of peptide-based HIV proteinase inhibitors. Science 1990, 248, 358–361. [Google Scholar] [CrossRef] [Green Version]
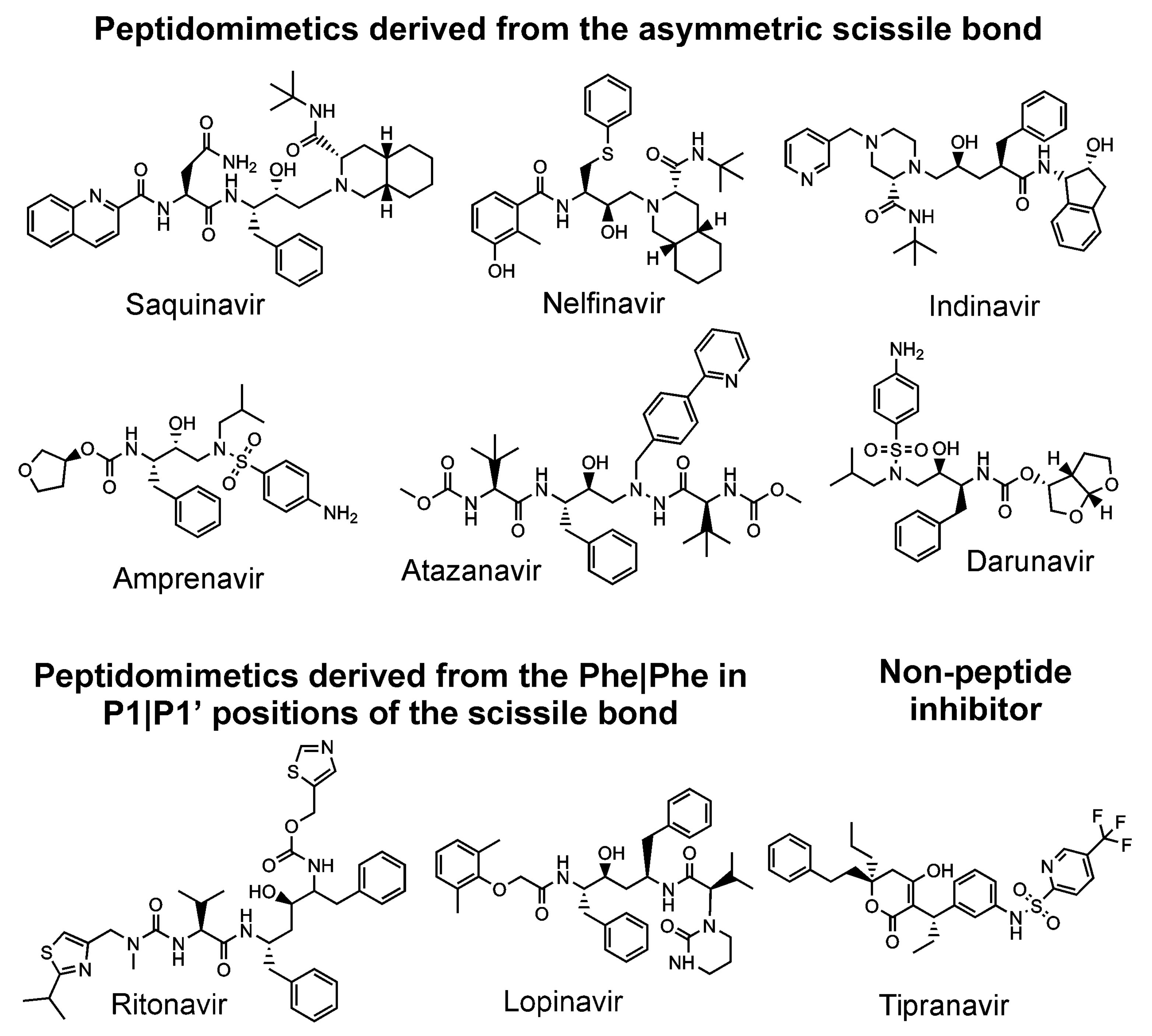
- Pokorná, J.; Machala, L.; Rezáčová, P.; Konvalinka, J. Current and Novel Inhibitors of HIV Protease. Viruses 2009, 1, 1209–1239. [Google Scholar] [CrossRef] [Green Version]
- Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284. [Google Scholar] [CrossRef] [Green Version]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef]
- Johns, B.A.; Kawasuji, T.; Weatherhead, J.G.; Taishi, T.; Temelkoff, D.P.; Yoshida, H.; Akiyama, T.; Taoda, Y.; Murai, H.; Kiyama, R.; et al. Carbamoyl pyridone HIV-1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J. Med. Chem. 2013, 56, 5901–5916. [Google Scholar] [CrossRef]
- Engelman, K.D.; Engelman, A.N. Long-Acting Cabotegravir for HIV/AIDS Prophylaxis. Biochemistry 2021, 60, 1731–1740. [Google Scholar] [CrossRef]
- Snásel, J.; Rosenberg, I.; Paces, O.; Pichová, I. The strand transfer oligonucleotide inhibitors of HIV-integrase. J. Enzym. Inhib. Med. Chem. 2009, 24, 241–246. [Google Scholar] [CrossRef]
- Hikichi, Y.; Van Duyne, R.; Pham, P.; Groebner, J.L.; Wiegand, A.; Mellors, J.W.; Kearney, M.F.; Freed, E.O. Mechanistic Analysis of the Broad Antiretroviral Resistance Conferred by HIV-1 Envelope Glycoprotein Mutations. mBio 2021, 12, e03134-20. [Google Scholar] [CrossRef]
- De Andrade Arrais, C.R.; Lima, K.; Barreiros, M.; Rodrigues, J.K.F.; Sousa, N.P.S.; Costa, D.D.; Santos, F.; Pereira, G.F.M.; AI, E.S.V.; Barros, A.K.; et al. HIV-1 subtypes and drug resistance in children during antiretroviral therapy in Brazil. J. Med. Virol. 2021, 93, 4908–4914. [Google Scholar] [CrossRef]
- Cilento, M.E.; Kirby, K.A.; Sarafianos, S.G. Avoiding Drug Resistance in HIV Reverse Transcriptase. Chem. Rev. 2021, 121, 3271–3296. [Google Scholar] [CrossRef]
- Nka, A.D.; Teto, G.; Santoro, M.M.; Ngum Ndze, V.; Takou, D.; Dambaya, B.; Ngoufack Jagni Semengue, E.; Fabeni, L.; Perno, C.F.; Colizzi, V.; et al. HIV-1 Gag gene mutations, treatment response and drug resistance to protease inhibitors: A systematic review and meta-analysis protocol. PLoS ONE 2021, 16, e0253587. [Google Scholar] [CrossRef]
- Agniswamy, J.; Kneller, D.W.; Ghosh, A.K.; Weber, I.T. Novel HIV PR inhibitors with C4-substituted bis-THF and bis-fluoro-benzyl target the two active site mutations of highly drug resistant mutant PR(S17). Biochem. Biophys. Res. Commun. 2021, 566, 30–35. [Google Scholar] [CrossRef]
- Weber, I.T.; Wang, Y.F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef]
- Kozísek, M.; Henke, S.; Sasková, K.G.; Jacobs, G.B.; Schuch, A.; Buchholz, B.; Müller, V.; Kräusslich, H.G.; Rezácová, P.; Konvalinka, J.; et al. Mutations in HIV-1 gag and pol compensate for the loss of viral fitness caused by a highly mutated protease. Antimicrob. Agents Chemother. 2012, 56, 4320–4330. [Google Scholar] [CrossRef] [Green Version]
- Fun, A.; van Maarseveen, N.M.; Pokorná, J.; Maas, R.E.; Schipper, P.J.; Konvalinka, J.; Nijhuis, M. HIV-1 protease inhibitor mutations affect the development of HIV-1 resistance to the maturation inhibitor bevirimat. Retrovirology 2011, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Sasková, K.G.; Kozísek, M.; Rezácová, P.; Brynda, J.; Yashina, T.; Kagan, R.M.; Konvalinka, J. Molecular characterization of clinical isolates of human immunodeficiency virus resistant to the protease inhibitor darunavir. J. Virol. 2009, 83, 8810–8818. [Google Scholar] [CrossRef] [Green Version]
- Mulato, A.; Acosta, R.; Chang, S.; Martin, R.; Yant, S.R.; Cihlar, T.; White, K. Simulating HIV Breakthrough and Resistance Development During Variable Adherence to Antiretroviral Treatment. J. Acquir. Immune Defic. Syndr. 2021, 86, 369–377. [Google Scholar] [CrossRef]
- Borghetti, A.; Ciccullo, A.; Lombardi, F.; Baldin, G.; Belmonti, S.; Prosperi, M.; Incardona, F.; Heger, E.; Borghi, V.; Sönnerborg, A.; et al. Transmitted drug resistance to NRTIs and risk of virological failure in naïve patients treated with integrase inhibitors. HIV Med. 2021, 22, 22–27. [Google Scholar] [CrossRef]
- Berríos-Caro, E.; Gifford, D.R.; Galla, T. Competition delays multi-drug resistance evolution during combination therapy. J. Theor. Biol. 2021, 509, 110524. [Google Scholar] [CrossRef]
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A review of the toxicity of HIV medications. J. Med. Toxicol. 2014, 10, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Takamune, N.; Nirasawa, T.; Aoki, M.; Morishita, Y.; Das, D.; Koh, Y.; Ghosh, A.K.; Misumi, S.; Mitsuya, H. Dimerization of HIV-1 protease occurs through two steps relating to the mechanism of protease dimerization inhibition by darunavir. Proc. Natl. Acad. Sci. USA 2014, 111, 12234–12239. [Google Scholar] [CrossRef] [Green Version]
- Aoki, M.; Danish, M.L.; Aoki-Ogata, H.; Amano, M.; Ide, K.; Das, D.; Koh, Y.; Mitsuya, H. Loss of the protease dimerization inhibition activity of tipranavir (TPV) and its association with the acquisition of resistance to TPV by HIV-1. J. Virol. 2012, 86, 13384–13396. [Google Scholar] [CrossRef] [Green Version]
- Louis, J.M.; Aniana, A.; Weber, I.T.; Sayer, J.M. Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9072–9077. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.A.; Soule, E.E.; Davidoff, K.S.; Daniels, S.I.; Naiman, N.E.; Yarchoan, R. Activity of human immunodeficiency virus type 1 protease inhibitors against the initial autocleavage in Gag-Pol polyprotein processing. Antimicrob. Agents Chemother. 2012, 56, 3620–3628. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Sayer, J.M.; Aniana, A.; Yu, X.; Weber, I.T.; Harrison, R.W.; Louis, J.M. Binding of Clinical Inhibitors to a Model Precursor of a Rationally Selected Multidrug Resistant HIV-1 Protease Is Significantly Weaker Than That to the Released Mature Enzyme. Biochemistry 2016, 55, 2390–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humpolíčková, J.; Weber, J.; Starková, J.; Mašínová, E.; Günterová, J.; Flaisigová, I.; Konvalinka, J.; Majerová, T. Inhibition of the precursor and mature forms of HIV-1 protease as a tool for drug evaluation. Sci. Rep. 2018, 8, 10438. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, N.K. Direct renin inhibition and the kidney. Nat. Rev. Nephrol. 2010, 6, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-H.; Chang, Y.-C.; Agniswamy, J.; Harrison, R.W.; Weber, I.T. Conformational variation of an extreme drug resistant mutant of HIV protease. J. Mol. Graph. Model. 2015, 62, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chang, Y.-C.E.; Louis, J.M.; Wang, Y.-F.; Harrison, R.W.; Weber, I.T. Structures of Darunavir-Resistant HIV-1 Protease Mutant Reveal Atypical Binding of Darunavir to Wide Open Flaps. ACS Chem. Biol. 2014, 9, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, L.; Tien, C.; LaBarbera, D.V.; Chen, C. Targeting HIV-1 Protease Autoprocessing for High-throughput Drug Discovery and Drug Resistance Assessment. Sci. Rep. 2019, 9, 301. [Google Scholar] [CrossRef]
- Aoki, M.; Das, D.; Hayashi, H.; Aoki-Ogata, H.; Takamatsu, Y.; Ghosh, A.K.; Mitsuya, H.; Prasad, V.R.; Shafer, R.; Kovari, L. Mechanism of Darunavir (DRV)’s High Genetic Barrier to HIV-1 Resistance: A Key V32I Substitution in Protease Rarely Occurs, but Once It Occurs, It Predisposes HIV-1 To Develop DRV Resistance. mBio 2018, 9, e02425-17. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Balasubramanian, S.; Senapati, S. Understanding the mechanism of HIV-1 protease inhibition by monoclonal antibodies. J. Mol. Graph. Model. 2021, 103, 107826. [Google Scholar] [CrossRef]
- Bowman, M.J.; Byrne, S.; Chmielewski, J. Switching between allosteric and dimerization inhibition of HIV-1 protease. Chem. Biol. 2005, 12, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Pietrucci, F.; Vargiu, A.V.; Kranjc, A. HIV-1 Protease Dimerization Dynamics Reveals a Transient Druggable Binding Pocket at the Interface. Sci. Rep. 2015, 5, 18555. [Google Scholar] [CrossRef] [Green Version]
- Koh, Y.; Matsumi, S.; Das, D.; Amano, M.; Davis, D.A.; Li, J.; Leschenko, S.; Baldridge, A.; Shioda, T.; Yarchoan, R.; et al. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J. Biol. Chem. 2007, 282, 28709–28720. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Poorman, R.A.; Maggiora, L.L.; Heinrikson, R.L.; Kézdy, F.J. Dissociative inhibition of dimeric enzymes. Kinetic characterization of the inhibition of HIV-1 protease by its COOH-terminal tetrapeptide. J. Biol. Chem. 1991, 266, 15591–15594. [Google Scholar] [CrossRef]
- Uhlíková, T.; Konvalinka, J.; Pichová, I.; Soucek, M.; Kräusslich, H.G.; Vondrásek, J. A modular approach to HIV-1 proteinase inhibitor design. Biochem. Biophys. Res. Commun. 1996, 222, 38–43. [Google Scholar] [CrossRef]
- Strisovsky, K.; Tessmer, U.; Langner, J.; Konvalinka, J.; Kräusslich, H.G. Systematic mutational analysis of the active-site threonine of HIV-1 proteinase: Rethinking the “fireman’s grip” hypothesis. Protein Sci. 2000, 9, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Ingr, M.; Uhlíková, T.; Strísovský, K.; Majerová, E.; Konvalinka, J. Kinetics of the dimerization of retroviral proteases: The “fireman’s grip” and dimerization. Protein Sci. 2003, 12, 2173–2182. [Google Scholar] [CrossRef]
- Shehu-Xhilaga, M.; Crowe, S.M.; Mak, J. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 2001, 75, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
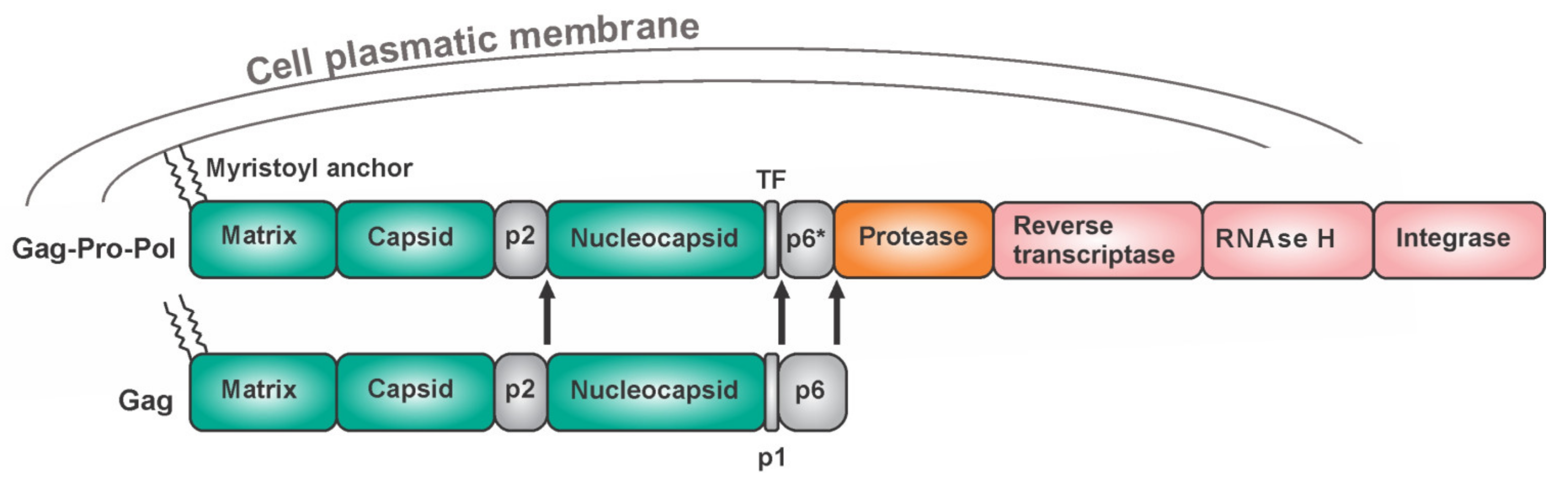
- Pettit, S.C.; Everitt, L.E.; Choudhury, S.; Dunn, B.M.; Kaplan, A.H. Initial cleavage of the human immunodeficiency virus type 1 GagPol precursor by its activated protease occurs by an intramolecular mechanism. J. Virol. 2004, 78, 8477–8485. [Google Scholar] [CrossRef] [Green Version]
- Pettit, S.C.; Clemente, J.C.; Jeung, J.A.; Dunn, B.M.; Kaplan, A.H. Ordered processing of the human immunodeficiency virus type 1 GagPol precursor is influenced by the context of the embedded viral protease. J. Virol. 2005, 79, 10601–10607. [Google Scholar] [CrossRef] [Green Version]
- Pettit, S.C.; Gulnik, S.; Everitt, L.; Kaplan, A.H. The dimer interfaces of protease and extra-protease domains influence the activation of protease and the specificity of GagPol cleavage. J. Virol. 2003, 77, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Tien, C.; Huang, L.; Watanabe, S.M.; Speidel, J.T.; Carter, C.A.; Chen, C. Context-dependent autoprocessing of human immunodeficiency virus type 1 protease precursors. PLoS ONE 2018, 13, e0191372. [Google Scholar] [CrossRef] [Green Version]
- Zábranský, A.; Andreánsky, M.; Hrusková-Heidingsfeldová, O.; Havlícek, V.; Hunter, E.; Ruml, T.; Pichová, I. Three active forms of aspartic proteinase from Mason-Pfizer monkey virus. Virology 1998, 245, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Veverka, V.; Bauerová, H.; Zábranský, A.; Lang, J.; Ruml, T.; Pichová, I.; Hrabal, R. Three-dimensional structure of a monomeric form of a retroviral protease. J. Mol. Biol. 2003, 333, 771–780. [Google Scholar] [CrossRef]
- Khatib, F.; DiMaio, F.; Cooper, S.; Kazmierczyk, M.; Gilski, M.; Krzywda, S.; Zabranska, H.; Pichova, I.; Thompson, J.; Popović, Z.; et al. Crystal structure of a monomeric retroviral protease solved by protein folding game players. Nat. Struct. Mol. Biol. 2011, 18, 1175–1177. [Google Scholar] [CrossRef]
- Wosicki, S.; Kazmierczyk, M.; Gilski, M.; Zabranska, H.; Pichova, I.; Jaskolski, M. Crystal structures of inhibitor complexes of M-PMV protease with visible flap loops. Protein Sci. 2021, 30, 1258–1263. [Google Scholar] [CrossRef]
- Hrusková-Heidingsfeldová, O.; Andreansky, M.; Fábry, M.; Bláha, I.; Strop, P.; Hunter, E. Cloning, Bacterial Expression, and Characterization of the Mason-Pfizer Monkey Virus Proteinase. J. Biol. Chem. 1995, 270, 15053–15058. [Google Scholar] [CrossRef] [Green Version]
- Nijhuis, M.; van Maarseveen, N.M.; Lastere, S.; Schipper, P.; Coakley, E.; Glass, B.; Rovenska, M.; de Jong, D.; Chappey, C.; Goedegebuure, I.W.; et al. A novel substrate-based HIV-1 protease inhibitor drug resistance mechanism. PLoS Med. 2007, 4, e36. [Google Scholar] [CrossRef]
- Xue, B.; Mizianty, M.J.; Kurgan, L.; Uversky, V.N. Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Cell Mol. Life Sci. 2012, 69, 1211–1259. [Google Scholar] [CrossRef]
- Jochmans, D.; Anders, M.; Keuleers, I.; Smeulders, L.; Kräusslich, H.-G.; Kraus, G.; Müller, B. Selective killing of human immunodeficiency virus infected cells by non-nucleoside reverse transcriptase inhibitor-induced activation of HIV protease. Retrovirology 2010, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Sudo, S.; Haraguchi, H.; Hirai, Y.; Gatanaga, H.; Sakuragi, J.-I.; Momose, F.; Morikawa, Y. Efavirenz enhances HIV-1 gag processing at the plasma membrane through Gag-Pol dimerization. J. Virol. 2013, 87, 3348–3360. [Google Scholar] [CrossRef] [Green Version]
- Rumlová, M.; Křížová, I.; Keprová, A.; Hadravová, R.; Doležal, M.; Strohalmová, K.; Pichová, I.; Hájek, M.; Ruml, T. HIV-1 protease-induced apoptosis. Retrovirology 2014, 11, 37. [Google Scholar] [CrossRef]
- Buzon, M.J.; Erkizia, I.; Pou, C.; Minuesa, G.; Puertas, M.C.; Esteve, A.; Castello, A.; Santos, J.R.; Prado, J.G.; Izquierdo-Useros, N.; et al. A non-infectious cell-based phenotypic assay for the assessment of HIV-1 susceptibility to protease inhibitors. J. Antimicrob. Chemother. 2012, 67, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, K.; Uhlíková, T.; Konvalinka, J.; Masucci, M.G.; Dantuma, N.P. Cell-based fluorescence assay for human immunodeficiency virus type 1 protease activity. Antimicrob. Agents Chemother. 2001, 45, 2616–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majerová-Uhlíková, T.; Dantuma, N.P.; Lindsten, K.; Masucci, M.G.; Konvalinka, J. Non-infectious fluorimetric assay for phenotyping of drug-resistant HIV proteinase mutants. J. Clin. Virol. 2006, 36, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.H.; Swanstrom, R. Human immunodeficiency virus type 1 Gag proteins are processed in two cellular compartments. Proc. Natl. Acad. Sci. USA 1991, 88, 4528–4532. [Google Scholar] [CrossRef] [Green Version]
- Trinité, B.; Zhang, H.; Levy, D.N. NNRTI-induced HIV-1 protease-mediated cytotoxicity induces rapid death of CD4 T cells during productive infection and latency reversal. Retrovirology 2019, 16, 17. [Google Scholar] [CrossRef] [Green Version]
- Jurado, K.A.; Engelman, A. Multimodal mechanism of action of allosteric HIV-1 integrase inhibitors. Expert Rev. Mol. Med. 2013, 15, e14. [Google Scholar] [CrossRef] [Green Version]
- Kräusslich, H.G. Human immunodeficiency virus proteinase dimer as component of the viral polyprotein prevents particle assembly and viral infectivity. Proc. Natl. Acad. Sci. USA 1991, 88, 3213–3217. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.Y.; Wang, S.M.; Huang, K.J.; Chiang, C.C.; Wang, C.T. Placement of leucine zipper motifs at the carboxyl terminus of HIV-1 protease significantly reduces virion production. PLoS ONE 2012, 7, e32845. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.C.; Wang, F.D.; Chen, Y.A.; Wang, C.T. Effects of human immunodeficiency virus type 1 transframe protein p6* mutations on viral protease-mediated Gag processing. J. Gen. Virol. 2006, 87, 2041–2046. [Google Scholar] [CrossRef]
- Zábranský, A.; Hadravová, R.; Stokrová, J.; Sakalian, M.; Pichová, I. Premature processing of mouse mammary tumor virus Gag polyprotein impairs intracellular capsid assembly. Virology 2009, 384, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Schur, F.K.; Hagen, W.J.; Rumlová, M.; Ruml, T.; Müller, B.; Kräusslich, H.G.; Briggs, J.A. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature 2015, 517, 505–508. [Google Scholar] [CrossRef]
- Bohmová, K.; Hadravová, R.; Stokrová, J.; Tuma, R.; Ruml, T.; Pichová, I.; Rumlová, M. Effect of dimerizing domains and basic residues on in vitro and in vivo assembly of Mason-Pfizer monkey virus and human immunodeficiency virus. J. Virol. 2010, 84, 1977–1988. [Google Scholar] [CrossRef] [Green Version]
- Junková, P.; Pleskot, R.; Prchal, J.; Sýs, J.; Ruml, T. Differences and commonalities in plasma membrane recruitment of the two morphogenetically distinct retroviruses HIV-1 and MMTV. J. Biol. Chem. 2020, 295, 8819–8833. [Google Scholar] [CrossRef]
- Kulkarni, M.M.; Ratcliff, A.N.; Bhat, M.; Alwarawrah, Y.; Hughes, P.; Arcos, J.; Loiselle, D.; Torrelles, J.B.; Funderburg, N.T.; Haystead, T.A.; et al. Cellular fatty acid synthase is required for late stages of HIV-1 replication. Retrovirology 2017, 14, 45. [Google Scholar] [CrossRef] [Green Version]
- Lindwasser, O.W.; Resh, M.D. Myristoylation as a target for inhibiting HIV assembly: Unsaturated fatty acids block viral budding. Proc. Natl. Acad. Sci. USA 2002, 99, 13037–13042. [Google Scholar] [CrossRef] [Green Version]
- Zell, R. Picornaviridae-the ever-growing virus family. Arch. Virol. 2018, 163, 299–317. [Google Scholar] [CrossRef]
- Sárkány, Z.; Polgár, L. The unusual catalytic triad of poliovirus protease 3C. Biochemistry 2003, 42, 516–522. [Google Scholar] [CrossRef]
- Horova, V.; Lyoo, H.; Różycki, B.; Chalupska, D.; Smola, M.; Humpolickova, J.; Strating, J.R.P.M.; van Kuppeveld, F.J.M.; Boura, E.; Klima, M. Convergent evolution in the mechanisms of ACBD3 recruitment to picornavirus replication sites. PLoS Pathog. 2019, 15, e1007962. [Google Scholar] [CrossRef] [Green Version]
- Parsley, T.B.; Cornell, C.T.; Semler, B.L. Modulation of the RNA binding and protein processing activities of poliovirus polypeptide 3CD by the viral RNA polymerase domain. J. Biol. Chem. 1999, 274, 12867–12876. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, L.L.; Wass, A.B.; Gohara, D.W.; Pathak, H.B.; Arnold, J.J.; Filman, D.J.; Cameron, C.E.; Hogle, J.M. Crystal structure of poliovirus 3CD protein: Virally encoded protease and precursor to the RNA-dependent RNA polymerase. J. Virol. 2007, 81, 3583–3596. [Google Scholar] [CrossRef] [Green Version]
- Winston, D.S.; Boehr, D.D. The Picornavirus Precursor 3CD Has Different Conformational Dynamics Compared to 3C(pro) and 3D(pol) in Functionally Relevant Regions. Viruses 2021, 13, 442. [Google Scholar] [CrossRef]
- Spear, A.; Ogram, S.A.; Morasco, B.J.; Smerage, L.E.; Flanegan, J.B. Viral precursor protein P3 and its processed products perform discrete and essential functions in the poliovirus RNA replication complex. Virology 2015, 485, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.M.; Moustafa, I.M.; Arnold, J.J.; Cameron, C.E.; Boehr, D.D. Long-Range Communication between Different Functional Sites in the Picornaviral 3C Protein. Structure 2016, 24, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Meng, B.; Lan, K.; Xie, J.; Lerner, R.A.; Wilson, I.A.; Yang, B. Inhibitory antibodies identify unique sites of therapeutic vulnerability in rhinovirus and other enteroviruses. Proc. Natl. Acad. Sci. USA 2020, 117, 13499–13508. [Google Scholar] [CrossRef]
- Rahnefeld, A.; Klingel, K.; Schuermann, A.; Diny, N.L.; Althof, N.; Lindner, A.; Bleienheuft, P.; Savvatis, K.; Respondek, D.; Opitz, E.; et al. Ubiquitin-like protein ISG15 (interferon-stimulated gene of 15 kDa) in host defense against heart failure in a mouse model of virus-induced cardiomyopathy. Circulation 2014, 130, 1589–1600. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, E.M.; James, M.N.G. The 3C proteinases of picornaviruses. In Proteases as Targets for Therapy; von der Helm, K., Korant, B.D., Cheronis, J.C., Eds.; Springer: Berlin/Heidelberg, Germany, 2000; pp. 117–143. [Google Scholar]
- Green, K.Y.; Kaufman, S.S.; Nagata, B.M.; Chaimongkol, N.; Kim, D.Y.; Levenson, E.A.; Tin, C.M.; Yardley, A.B.; Johnson, J.A.; Barletta, A.B.F.; et al. Human norovirus targets enteroendocrine epithelial cells in the small intestine. Nat. Commun. 2020, 11, 2759. [Google Scholar] [CrossRef]
- Muzzarelli, K.M.; Kuiper, B.; Spellmon, N.; Brunzelle, J.; Hackett, J.; Amblard, F.; Zhou, S.; Liu, P.; Kovari, I.A.; Yang, Z.; et al. Structural and Antiviral Studies of the Human Norovirus GII.4 Protease. Biochemistry 2019, 58, 900–907. [Google Scholar] [CrossRef]
- Viskovska, M.A.; Zhao, B.; Shanker, S.; Choi, J.-M.; Deng, L.; Song, Y.; Palzkill, T.; Hu, L.; Estes, M.K.; Prasad, B.V.V.; et al. GII.4 Norovirus Protease Shows pH-Sensitive Proteolysis with a Unique Arg-His Pairing in the Catalytic Site. J. Virol. 2019, 93, e01479-18. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.O.; Kim, Y.; Lovell, S.; Rathnayake, A.D.; Groutas, W.C. Antiviral Drug Discovery: Norovirus Proteases and Development of Inhibitors. Viruses 2019, 11, 197. [Google Scholar] [CrossRef] [Green Version]
- Abu Bakar, F.; Ng, L.F.P. Nonstructural Proteins of Alphavirus—Potential Targets for Drug Development. Viruses 2018, 10, 71. [Google Scholar] [CrossRef] [Green Version]
- Chung, B.Y.; Firth, A.E.; Atkins, J.F. Frameshifting in alphaviruses: A diversity of 3’ stimulatory structures. J. Mol. Biol. 2010, 397, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Potužník, J.F.; Cahová, H. It’s the Little Things (in Viral RNA). mBio 2020, 11, e02131-20. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.J.; Hardy, W.R.; Shirako, Y.; Strauss, J.H. Cleavage-site preferences of Sindbis virus polyproteins containing the non-structural proteinase. Evidence for temporal regulation of polyprotein processing in vivo. EMBO J. 1990, 9, 2631–2638. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.; Bragagnolo, G.; Arranz, R.; Reguera, J. Capping pores of alphavirus nsP1 gate membranous viral replication factories. Nature 2021, 589, 615–619. [Google Scholar] [CrossRef]
- Hellström, K.; Kallio, K.; Utt, A.; Quirin, T.; Jokitalo, E.; Merits, A.; Ahola, T.; Simon, A.E. Partially Uncleaved Alphavirus Replicase Forms Spherule Structures in the Presence and Absence of RNA Template. J. Virol. 2017, 91, e00787-17. [Google Scholar] [CrossRef] [Green Version]
- Shirako, Y.; Strauss, J.H. Regulation of Sindbis virus RNA replication: Uncleaved P123 and nsP4 function in minus-strand RNA synthesis, whereas cleaved products from P123 are required for efficient plus-strand RNA synthesis. J. Virol. 1994, 68, 1874–1885. [Google Scholar] [CrossRef] [Green Version]
- Lemm, J.A.; Rümenapf, T.; Strauss, E.G.; Strauss, J.H.; Rice, C.M. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: A model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J. 1994, 13, 2925–2934. [Google Scholar] [CrossRef] [Green Version]
- Shin, G.; Yost, S.A.; Miller, M.T.; Elrod, E.J.; Grakoui, A.; Marcotrigiano, J. Structural and functional insights into alphavirus polyprotein processing and pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 16534–16539. [Google Scholar] [CrossRef] [Green Version]
- Lulla, A.; Lulla, V.; Tints, K.; Ahola, T.; Merits, A. Molecular determinants of substrate specificity for Semliki Forest virus nonstructural protease. J. Virol. 2006, 80, 5413–5422. [Google Scholar] [CrossRef] [Green Version]
- Lulla, V.; Karo-Astover, L.; Rausalu, K.; Saul, S.; Merits, A.; Lulla, A. Timeliness of Proteolytic Events Is Prerequisite for Efficient Functioning of the Alphaviral Replicase. J. Virol. 2018, 92, e00151-18. [Google Scholar] [CrossRef] [Green Version]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef]
- Castle, E.; Nowak, T.; Leidner, U.; Wengler, G.; Wengler, G. Sequence analysis of the viral core protein and the membrane-associated proteins V1 and NV2 of the flavivirus west nile virus and of the genome sequence for these proteins. Virology 1985, 145, 227–236. [Google Scholar] [CrossRef]
- Nowak, T.; Färber, P.M.; Wengler, G.; Wengler, G. Analyses of the terminal sequences of west nile virus structural proteins and of the in vitro translation of these proteins allow the proposal of a complete scheme of the proteolytic cleavages involved in their synthesis. Virology 1989, 169, 365–376. [Google Scholar] [CrossRef]
- Falgout, B.; Pethel, M.; Zhang, Y.M.; Lai, C.J. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 1991, 65, 2467–2475. [Google Scholar] [CrossRef] [Green Version]
- Amberg, S.M.; Nestorowicz, A.; McCourt, D.W.; Rice, C.M. NS2B-3 proteinase-mediated processing in the yellow fever virus structural region: In vitro and in vivo studies. J. Virol. 1994, 68, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Rana, J.; Burrone, O.R. DENV2 Pseudoviral Particles with Unprocessed Capsid Protein Are Assembled and Infectious. Viruses 2019, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Kurz, M.; Stefan, N.; Zhu, J.; Skern, T. NS2B/3 proteolysis at the C-prM junction of the tick-borne encephalitis virus polyprotein is highly membrane dependent. Virus Res. 2012, 168, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Stadler, K.; Allison, S.L.; Schalich, J.; Heinz, F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997, 71, 8475–8481. [Google Scholar] [CrossRef] [Green Version]
- Yu, I.-M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the Immature Dengue Virus at Low pH Primes Proteolytic Maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef]
- Plevka, P.; Battisti, A.J.; Sheng, J.; Rossmann, M.G. Mechanism for maturation-related reorganization of flavivirus glycoproteins. J. Struct. Biol. 2014, 185, 27–31. [Google Scholar] [CrossRef] [Green Version]
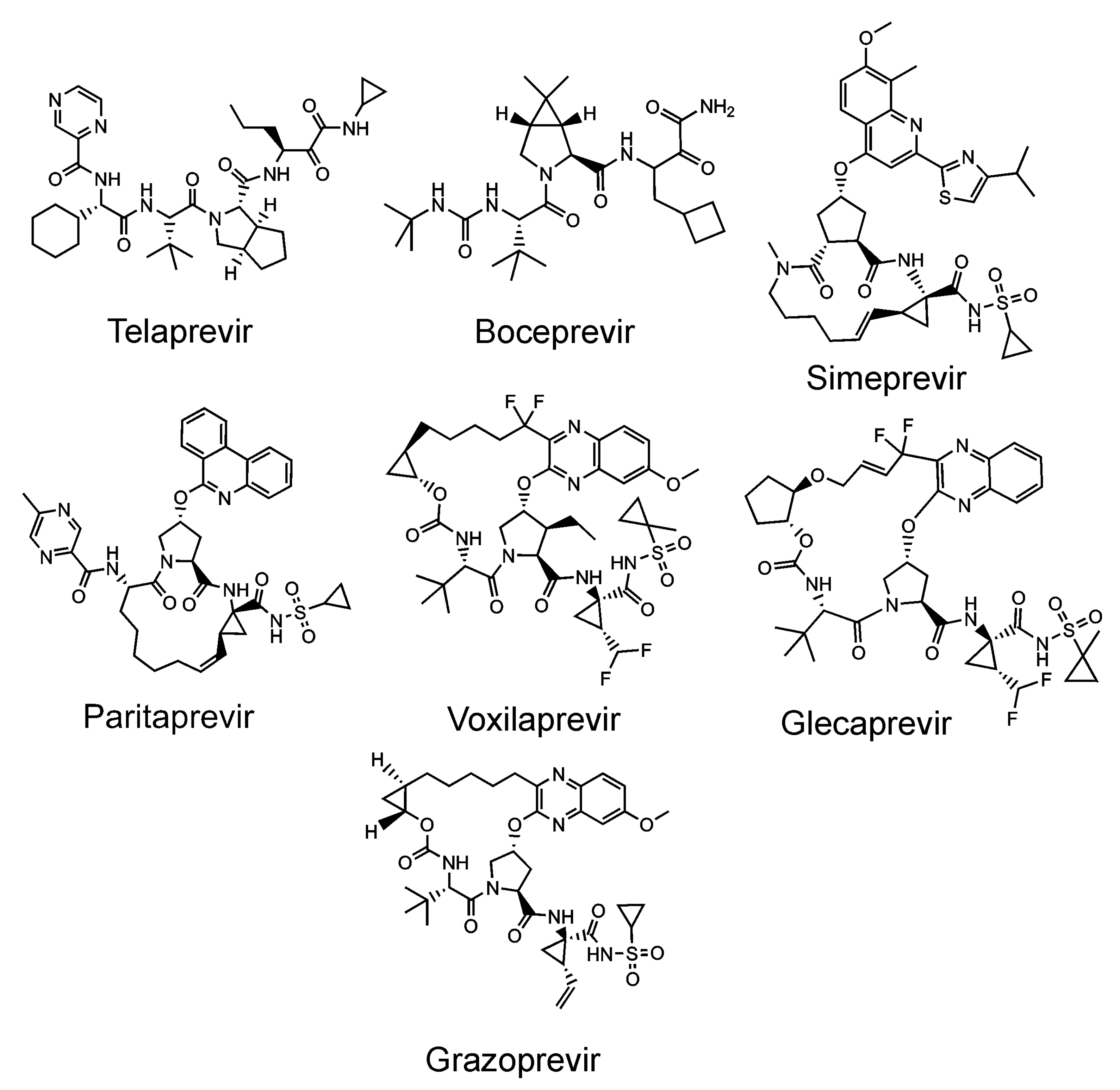
- Lin, C.; Rice, C.M. The hepatitis C virus NS3 serine proteinase and NS4A cofactor: Establishment of a cell-free trans-processing assay. Proc. Natl. Acad. Sci. USA 1995, 92, 7622–7626. [Google Scholar] [CrossRef] [Green Version]
- Clark, V.C.; Peter, J.A.; Nelson, D.R. New therapeutic strategies in HCV: Second-generation protease inhibitors. Liver Int. 2013, 33 Suppl 1, 80–84. [Google Scholar] [CrossRef]
- Majerová, T.; Novotný, P.; Krýsová, E.; Konvalinka, J. Exploiting the unique features of Zika and Dengue proteases for inhibitor design. Biochimie 2019, 166, 132–141. [Google Scholar] [CrossRef]
- Li, Q.; Kang, C. Structure and Dynamics of Zika Virus Protease and Its Insights into Inhibitor Design. Biomedicines 2021, 9, 1044. [Google Scholar] [CrossRef]
- Gupta, G.; Lim, L.; Song, J. NMR and MD Studies Reveal That the Isolated Dengue NS3 Protease Is an Intrinsically Disordered Chymotrypsin Fold Which Absolutely Requests NS2B for Correct Folding and Functional Dynamics. PLoS ONE 2015, 10, e0134823. [Google Scholar] [CrossRef]
- Yusof, R.; Clum, S.; Wetzel, M.; Murthy, H.M.K.; Padmanabhan, R. Purified NS2B/NS3 Serine Protease of Dengue Virus Type 2 Exhibits Cofactor NS2B Dependence for Cleavage of Substrates with Dibasic Amino Acids. J. Biol. Chem. 2000, 275, 9963–9969. [Google Scholar] [CrossRef] [Green Version]
- Brinkworth, R.I.; Fairlie, D.P.; Leung, D.; Young, P.R. Homology model of the dengue 2 virus NS3 protease: Putative interactions with both substrate and NS2B cofactor. J. Gen. Virol. 1999, 80, 1167–1177. [Google Scholar] [CrossRef]
- Leung, D.; Schroder, K.; White, H.; Fang, N.-X.; Stoermer, M.J.; Abbenante, G.; Martin, J.L.; Young, P.R.; Fairlie, D.P. Activity of Recombinant Dengue 2 Virus NS3 Protease in the Presence of a Truncated NS2B Co-factor, Small Peptide Substrates, and Inhibitors. J. Biol. Chem. 2001, 276, 45762–45771. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef] [Green Version]
- Behnam, M.A.M.; Klein, C.D.P. Conformational selection in the flaviviral NS2B-NS3 protease. Biochimie 2020, 174, 117–125. [Google Scholar] [CrossRef]
- Mastrangelo, E.; Milani, M.; Bollati, M.; Selisko, B.; Peyrane, F.; Pandini, V.; Sorrentino, G.; Canard, B.; Konarev, P.V.; Svergun, D.I.; et al. Crystal Structure and Activity of Kunjin Virus NS3 Helicase; Protease and Helicase Domain Assembly in the Full Length NS3 Protein. J. Mol. Biol. 2007, 372, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Sampath, A.; Chao, A.; Wen, D.; Nanao, M.; Chene, P.; Vasudevan, S.G.; Lescar, J. Structure of the Dengue Virus Helicase/Nucleoside Triphosphatase Catalytic Domain at a Resolution of 2.4 A. J. Virol. 2005, 79, 10278–10288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, D.; Wei, N.; Doan, D.N.; Paradkar, P.N.; Chong, Y.; Davidson, A.D.; Kotaka, M.; Lescar, J.; Vasudevan, S.G. Flexibility between the protease and helicase domains of the dengue virus NS3 protein conferred by the linker region and its functional implications. J. Biol. Chem. 2010, 285, 18817–18827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Huo, T.; Lin, Y.L.; Nie, S.; Wu, F.; Hua, Y.; Wu, J.; Kneubehl, A.R.; Vogt, M.B.; Rico-Hesse, R.; et al. Discovery, X-ray Crystallography and Antiviral Activity of Allosteric Inhibitors of Flavivirus NS2B-NS3 Protease. J. Am. Chem. Soc. 2019, 141, 6832–6836. [Google Scholar] [CrossRef]
- Teo, K.F.; Wright, P.J. Internal proteolysis of the NS3 protein specified by dengue virus 2. J. Gen. Virol. 1997, 78, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Kümmerer, B.M.; Rice, C.M. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 2002, 76, 4773–4784. [Google Scholar] [CrossRef] [Green Version]
- Constant, D.A.; Mateo, R.; Nagamine, C.M.; Kirkegaard, K. Targeting intramolecular proteinase NS2B/3 cleavages for trans-dominant inhibition of dengue virus. Proc. Natl. Acad. Sci. USA 2018, 115, 10136–10141. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.J.H.; Lee, R.C.H.; Ang, M.J.Y.; Wang, W.-L.; Lim, H.A.; Wee, J.L.K.; Joy, J.; Hill, J.; Brian Chia, C.S. Antiviral activities of 15 dengue NS2B-NS3 protease inhibitors using a human cell-based viral quantification assay. Antivir. Res. 2015, 118, 68–74. [Google Scholar] [CrossRef]
- Tomlinson, S.M.; Watowich, S.J. Anthracene-based inhibitors of dengue virus NS2B–NS3 protease. Antivir. Res. 2011, 89, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Swarbrick, C.; Zogali, V.; Chan, K.W.K.; Kiousis, D.; Gwee, C.P.; Wang, S.; Lescar, J.; Luo, D.; von Itzstein, M.; Matsoukas, M.-T.; et al. Amidoxime prodrugs convert to potent cell-active multimodal inhibitors of the dengue virus protease. Eur. J. Med. Chem. 2021, 224, 113695. [Google Scholar] [CrossRef]
- Richter, M.; Leuthold, M.M.; Graf, D.; Bartenschlager, R.; Klein, C.D. Prodrug Activation by a Viral Protease: Evaluating Combretastatin Peptide Hybrids to Selectively Target Infected Cells. ACS Med. Chem. Lett. 2019, 10, 1115–1121. [Google Scholar] [CrossRef]
- Luo, D.; Xu, T.; Hunke, C.; Grüber, G.; Vasudevan, S.G.; Lescar, J. Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 2008, 82, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Li, K.S.M.; Poon, R.W.S.; Wong, B.H.L.; Tsoi, H.-W.; Yip, B.C.K.; Huang, Y.; Chan, K.-H.; Yuen, K.-Y. Molecular diversity of coronaviruses in bats. Virology 2006, 351, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Liya, G.; Yuguang, W.; Jian, L.; Huaiping, Y.; Xue, H.; Jianwei, H.; Jiaju, M.; Youran, L.; Chen, M.; Yiqing, J. Studies on viral pneumonia related to novel coronavirus SARS-CoV-2, SARS-CoV, and MERS-CoV: A literature review. APMIS 2020, 128, 423–432. [Google Scholar] [CrossRef]
- Spaan, W.; Cavanagh, D.; Horzinek, M.C. Coronaviruses: Structure and Genome Expression. J. Gen. Virol. 1988, 69, 2939–2952. [Google Scholar] [CrossRef]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.-h.; et al. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [Green Version]
- Ratia, K.; Saikatendu, K.S.; Santarsiero, B.D.; Barretto, N.; Baker, S.C.; Stevens, R.C.; Mesecar, A.D. Severe acute respiratory syndrome coronavirus papain-like protease: Structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. USA 2006, 103, 5717–5722. [Google Scholar] [CrossRef] [Green Version]
- Ziebuhr, J.; Herold, J.; Siddell, S.G. Characterization of a human coronavirus (strain 229E) 3C-like proteinase activity. J. Virol. 1995, 69, 4331–4338. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, P.R.; Scaiola, A.; Loughran, G.; Leibundgut, M.; Kratzel, A.; Meurs, R.; Dreos, R.; O’Connor, K.M.; McMillan, A.; Bode, J.W.; et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science 2021, 372, 1306–1313. [Google Scholar] [CrossRef]
- Grum-Tokars, V.; Ratia, K.; Begaye, A.; Baker, S.C.; Mesecar, A.D. Evaluating the 3C-like protease activity of SARS-Coronavirus: Recommendations for standardized assays for drug discovery. Virus Res. 2008, 133, 63–73. [Google Scholar] [CrossRef]
- Zhang, S.; Zhong, N.; Xue, F.; Kang, X.; Ren, X.; Chen, J.; Jin, C.; Lou, Z.; Xia, B. Three-dimensional domain swapping as a mechanism to lock the active conformation in a super-active octamer of SARS-CoV main protease. Protein Cell 2010, 1, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wei, P.; Huang, C.; Tan, L.; Liu, Y.; Lai, L. Only one protomer is active in the dimer of SARS 3C-like proteinase. J. Biol. Chem. 2006, 281, 13894–13898. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Jaskolski, M.; Dauter, Z.; Shabalin, I.G.; Gilski, M.; Brzezinski, D.; Kowiel, M.; Rupp, B.; Wlodawer, A. Crystallographic models of SARS-CoV-2 3CLpro: In-depth assessment of structure quality and validation. IUCrJ 2021, 8, 238–256. [Google Scholar] [CrossRef] [PubMed]
- Jaffrelot Inizan, T.; Célerse, F.; Adjoua, O.; El Ahdab, D.; Jolly, L.H.; Liu, C.; Ren, P.; Montes, M.; Lagarde, N.; Lagardère, L.; et al. High-resolution mining of the SARS-CoV-2 main protease conformational space: Supercomputer-driven unsupervised adaptive sampling. Chem. Sci. 2021, 12, 4889–4907. [Google Scholar] [CrossRef] [PubMed]
- Barrila, J.; Bacha, U.; Freire, E. Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry 2006, 45, 14908–14916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, M.F.; Kuo, C.J.; Chang, K.T.; Chang, H.C.; Chou, C.C.; Ko, T.P.; Shr, H.L.; Chang, G.G.; Wang, A.H.; Liang, P.H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005, 280, 31257–31266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, T.; Kim, Y.-T.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. Autoprocessing mechanism of severe acute respiratory syndrome coronavirus 3C-like protease (SARS-CoV 3CLpro) from its polyproteins. FEBS J. 2013, 280, 2002–2013. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Jonas, F.; Shen, C.; Hilgenfeld, R. Liberation of SARS-CoV main protease from the viral polyprotein: N-terminal autocleavage does not depend on the mature dimerization mode. Protein Cell 2010, 1, 59–74. [Google Scholar] [CrossRef]
- Noske, G.D.; Nakamura, A.M.; Gawriljuk, V.O.; Fernandes, R.S.; Lima, G.M.A.; Rosa, H.V.D.; Pereira, H.D.; Zeri, A.C.M.; Nascimento, A.F.Z.; Freire, M.C.L.C.; et al. A Crystallographic Snapshot of SARS-CoV-2 Main Protease Maturation Process. J. Mol. Biol. 2021, 433, 167118. [Google Scholar] [CrossRef]
- Roe, M.K.; Junod, N.A.; Young, A.R.; Beachboard, D.C.; Stobart, C.C. Targeting novel structural and functional features of coronavirus protease nsp5 (3CLpro, Mpro) in the age of COVID-19. J. Gen. Virol. 2021, 102, 1558. [Google Scholar] [CrossRef]
- Lee, J.; Worrall, L.J.; Vuckovic, M.; Rosell, F.I.; Gentile, F.; Ton, A.-T.; Caveney, N.A.; Ban, F.; Cherkasov, A.; Paetzel, M.; et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 2020, 11, 5877. [Google Scholar] [CrossRef]
- Fan, K.; Wei, P.; Feng, Q.; Chen, S.; Huang, C.; Ma, L.; Lai, B.; Pei, J.; Liu, Y.; Chen, J.; et al. Biosynthesis, Purification, and Substrate Specificity of Severe Acute Respiratory Syndrome Coronavirus 3C-like Proteinase. J. Biol. Chem. 2004, 279, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Sivaraman, J.; Song, J. Mechanism for Controlling the Dimer-Monomer Switch and Coupling Dimerization to Catalysis of the Severe Acute Respiratory Syndrome Coronavirus 3C-Like Protease. J. Virol. 2008, 82, 4620–4629. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-C.; Chang, G.-G.; Chou, C.-Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 2010, 98, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Muhaxhiri, Z.; Deng, L.; Shanker, S.; Sankaran, B.; Estes, M.K.; Palzkill, T.; Song, Y.; Prasad, B.V.V. Structural basis of substrate specificity and protease inhibition in Norwalk virus. J. Virol. 2013, 87, 4281–4292. [Google Scholar] [CrossRef] [Green Version]
- Ingr, M.; Lange, R.; Halabalová, V.; Yehya, A.; Hrnčiřík, J.; Chevalier-Lucia, D.; Palmade, L.; Blayo, C.; Konvalinka, J.; Dumay, E. Inhibitor and substrate binding induced stability of HIV-1 protease against sequential dissociation and unfolding revealed by high pressure spectroscopy and kinetics. PLoS ONE 2015, 10, e0119099. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Song, J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef]
- Lim, L.; Gupta, G.; Roy, A.; Kang, J.; Srivastava, S.; Shi, J.; Song, J. Structurally- and dynamically-driven allostery of the chymotrypsin-like proteases of SARS, Dengue and Zika viruses. Prog. Biophys. Mol. Biol. 2019, 143, 52–66. [Google Scholar] [CrossRef]
- Kokkonen, P.; Slanska, M.; Dockalova, V.; Pinto, G.P.; Sánchez-Carnerero, E.M.; Damborsky, J.; Klán, P.; Prokop, Z.; Bednar, D. The impact of tunnel mutations on enzymatic catalysis depends on the tunnel-substrate complementarity and the rate-limiting step. Comput. Struct. Biotechnol. J. 2020, 18, 805–813. [Google Scholar] [CrossRef]
- Rathnayake, A.D.; Zheng, J.; Kim, Y.; Perera, K.D.; Mackin, S.; Meyerholz, D.K.; Kashipathy, M.M.; Battaile, K.P.; Lovell, S.; Perlman, S.; et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV–infected mice. Sci. Transl. Med. 2020, 12, eabc5332. [Google Scholar] [CrossRef]
- Kim, Y.; Shivanna, V.; Narayanan, S.; Prior, A.M.; Weerasekara, S.; Hua, D.H.; Kankanamalage, A.C.G.; Groutas, W.C.; Chang, K.-O. Broad-spectrum inhibitors against 3C-like proteases of feline coronaviruses and feline caliciviruses. J. Virol. 2015, 89, 4942–4950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef]
- Vandyck, K.; Deval, J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr. Opin. Virol. 2021, 49, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Reinke, P.Y.A.; Fernández-García, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.S.; Hui, K.P.Y.; Lai, H.M.; He, X.; Khan, K.S.; Kaur, S.; Huang, J.; Li, Z.; Chan, A.K.N.; Cheung, H.H.; et al. Simeprevir Potently Suppresses SARS-CoV-2 Replication and Synergizes with Remdesivir. ACS Cent. Sci. 2021, 7, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Drayman, N.; DeMarco, J.K.; Jones, K.A.; Azizi, S.A.; Froggatt, H.M.; Tan, K.; Maltseva, N.I.; Chen, S.; Nicolaescu, V.; Dvorkin, S.; et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science 2021, 373, 931–936. [Google Scholar] [CrossRef]
- Tian, D.; Liu, Y.; Liang, C.; Xin, L.; Xie, X.; Zhang, D.; Wan, M.; Li, H.; Fu, X.; Liu, H.; et al. An update review of emerging small-molecule therapeutic options for COVID-19. Biomed. Pharmacother. 2021, 137, 111313. [Google Scholar] [CrossRef]
- Yang, H.; Yang, J. A review of the latest research on M(pro) targeting SARS-COV inhibitors. RSC Med. Chem. 2021, 12, 1026–1036. [Google Scholar] [CrossRef]
- El-Baba, T.J.; Lutomski, C.A.; Kantsadi, A.L.; Malla, T.R.; John, T.; Mikhailov, V.; Bolla, J.R.; Schofield, C.J.; Zitzmann, N.; Vakonakis, I.; et al. Allosteric Inhibition of the SARS-CoV-2 Main Protease: Insights from Mass Spectrometry Based Assays. Angew. Chem. Int. Ed. Engl. 2020, 59, 23544–23548. [Google Scholar] [CrossRef]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 2021, 12, 3623. [Google Scholar] [CrossRef]
- Rizzuti, B.; Grande, F.; Conforti, F.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Vega, S.; Reyburn, H.T.; Abian, O.; Velazquez-Campoy, A. Rutin Is a Low Micromolar Inhibitor of SARS-CoV-2 Main Protease 3CLpro: Implications for Drug Design of Quercetin Analogs. Biomedicines 2021, 9, 375. [Google Scholar] [CrossRef]
- Rizzuti, B.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Vega, S.; Grande, F.; Conforti, F.; Abian, O.; Velazquez-Campoy, A. Sub-Micromolar Inhibition of SARS-CoV-2 3CLpro by Natural Compounds. Pharmaceuticals 2021, 14, 892. [Google Scholar] [CrossRef]
- Peñalver, L.; Schmid, P.; Szamosvári, D.; Schildknecht, S.; Globisch, C.; Sawade, K.; Peter, C.; Böttcher, T.A. Ligand Selection Strategy Identifies Chemical Probes Targeting the Proteases of SARS-CoV-2. Angew. Chem. Int. Ed. 2021, 60, 6799–6806. [Google Scholar] [CrossRef]
- Kneller, D.W.; Galanie, S.; Phillips, G.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Malleability of the SARS-CoV-2 3CL M(pro) Active-Site Cavity Facilitates Binding of Clinical Antivirals. Structure 2020, 28, 1313–1320. [Google Scholar] [CrossRef]
- Behnam, M.A.M. Protein structural heterogeneity: A hypothesis for the basis of proteolytic recognition by the main protease of SARS-CoV and SARS-CoV-2. Biochimie 2021, 182, 177–184. [Google Scholar] [CrossRef]
- Mattei, S.; Anders, M.; Konvalinka, J.; Kräusslich, H.-G.; Briggs, J.A.G.; Müller, B. Induced maturation of human immunodeficiency virus. J. Virol. 2014, 88, 13722–13731. [Google Scholar] [CrossRef] [Green Version]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef] [Green Version]
- Kanjanahaluethai, A.; Baker, S.C. Identification of mouse hepatitis virus papain-like proteinase 2 activity. J. Virol. 2000, 74, 7911–7921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicki, S.G.; Sawicki, D.L.; Younker, D.; Meyer, Y.; Thiel, V.; Stokes, H.; Siddell, S.G. Functional and Genetic Analysis of Coronavirus Replicase-Transcriptase Proteins. PLoS Pathog. 2005, 1, e39. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The Papain-Like Protease of Severe Acute Respiratory Syndrome Coronavirus Has Deubiquitinating Activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Wang, Y.; Ratia, K.; Mesecar, A.D.; Wilkinson, K.D.; Baker, S.C. Proteolytic Processing and Deubiquitinating Activity of Papain-Like Proteases of Human Coronavirus NL63. J. Virol. 2007, 81, 6007–6018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wu, G. Spatial and temporal roles of SARS-CoV PLpro—A snapshot. FASEB J. 2021, 35, e21197. [Google Scholar] [CrossRef]
- Cihlova, B.; Huskova, A.; Böserle, J.; Nencka, R.; Boura, E.; Silhan, J. High-Throughput Fluorescent Assay for Inhibitor Screening of Proteases from RNA Viruses. Molecules 2021, 26, 3792. [Google Scholar] [CrossRef]
- Lin, M.H.; Moses, D.C.; Hsieh, C.H.; Cheng, S.C.; Chen, Y.H.; Sun, C.Y.; Chou, C.Y. Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antivir. Res. 2018, 150, 155–163. [Google Scholar] [CrossRef]
- Ara, A.; Kadoya, R.; Ishimura, H.; Shimamura, K.; Sylte, I.; Kurita, N. Specific interactions between zinc metalloproteinase anditsinhibitors: Ab initio fragment molecular orbital calculations. J. Mol. Graph. Model. 2017, 75, 277–286. [Google Scholar] [CrossRef]
- Armstrong, L.A.; Lange, S.M.; Dee Cesare, V.; Matthews, S.P.; Nirujogi, R.S.; Cole, I.; Hope, A.; Cunningham, F.; Toth, R.; Mukherjee, R.; et al. Biochemical characterization of protease activity of Nsp3 from SARS-CoV-2 and its inhibition bynanobodies. PLoS ONE 2021, 16, e0253364. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majerová, T.; Novotný, P. Precursors of Viral Proteases as Distinct Drug Targets. Viruses 2021, 13, 1981. https://doi.org/10.3390/v13101981
Majerová T, Novotný P. Precursors of Viral Proteases as Distinct Drug Targets. Viruses. 2021; 13(10):1981. https://doi.org/10.3390/v13101981
Chicago/Turabian StyleMajerová, Taťána, and Pavel Novotný. 2021. "Precursors of Viral Proteases as Distinct Drug Targets" Viruses 13, no. 10: 1981. https://doi.org/10.3390/v13101981
APA StyleMajerová, T., & Novotný, P. (2021). Precursors of Viral Proteases as Distinct Drug Targets. Viruses, 13(10), 1981. https://doi.org/10.3390/v13101981