
RNA-Seq Analysis of Influenza A Virus-Induced Transcriptional Changes in Mice Lung and Its Possible Implications for the Virus Pathogenicity in Mice

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Viral Propagation
2.2. Viral Infection
2.3. Library Construction and RNA Sequencing
2.4. Transcriptome Assembly and Reads Mapping
2.5. Expression and Function Annotation Analysis
2.6. GO Enrichment and KEGG Pathway Analysis
2.7. Gene Set Enrichment Analysis (GSEA)
2.8. Construction of a Scale-Free Co-Expression Network Using WGCNA
2.9. Validation of RNA-Seq Data
3. Results
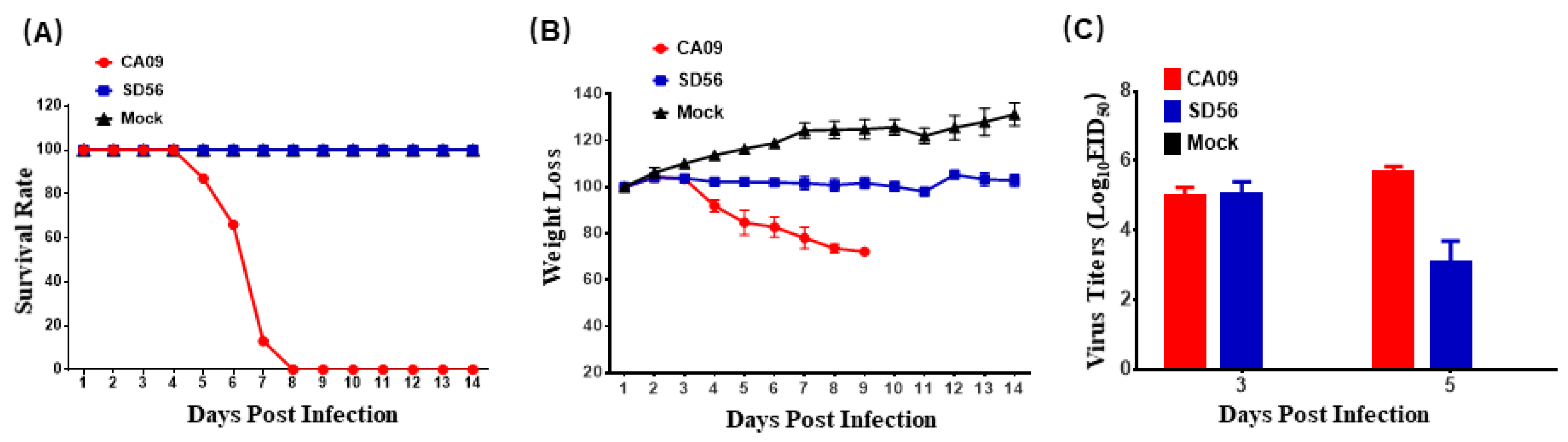
3.1. Pathogenicity of CA09 and SD56 in Mice
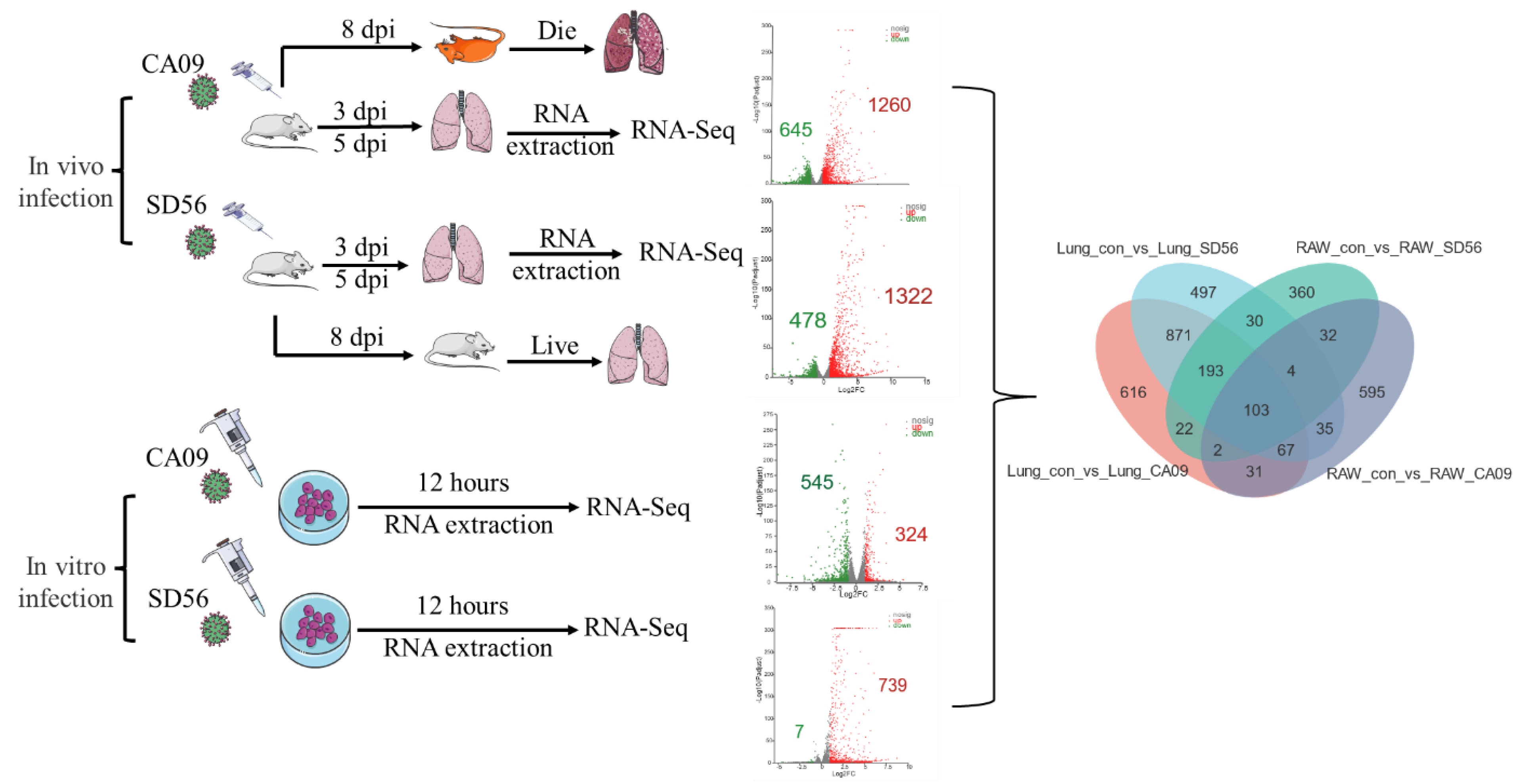
3.2. Global Transcriptome Changes Induced by IAV Infection in Lungs and RAW264.7 Cells
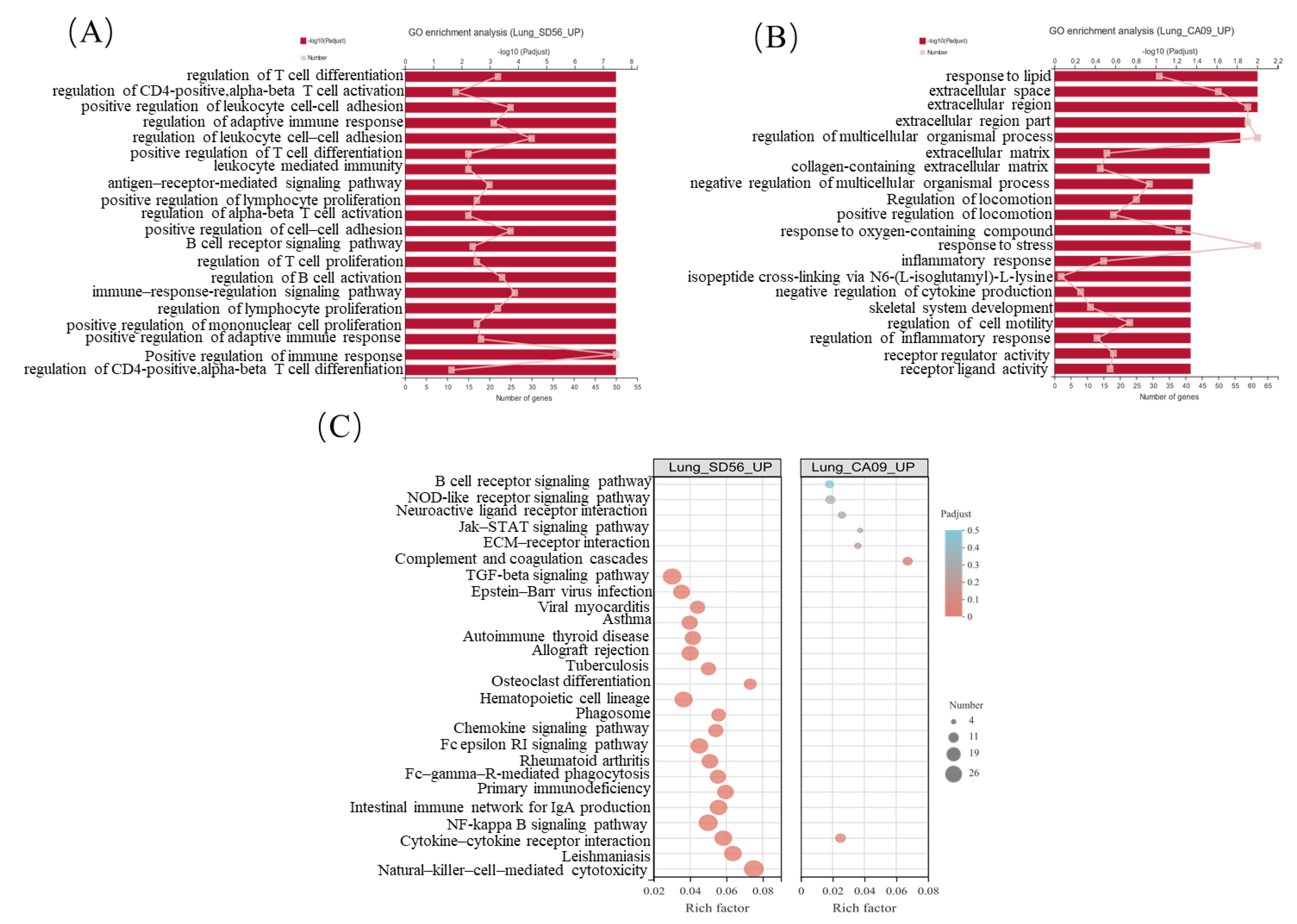
3.3. SD56 Infection Stimulated a Stronger Immune Response Than CA09 in Mice Lungs
3.4. CA09 Infection Significantly Regulated ECM-Related Pathways in Mouse Lungs
3.5. Abnormal Regulation of Macrophage Cell-Cycle-Related Genes after CA09 Infection
3.6. Weighted Gene Co-Expression Network Analysis Revealed the Hub Genes in CA09- and SD56-Infected Macrophages
3.7. Quantitative Real-Time PCR Validation of RNA-Seq Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Yoon, S.W.; Webby, R.J.; Webster, R.G. Evolution and ecology of influenza A viruses. Curr. Top. Microbiol. Immunol. 2014, 385, 359–375. [Google Scholar]
- Francis, M.E.; King, M.L.; Kelvin, A.A. Back to the future for influenza preimmunity-looking back at influenza virus history to infer the outcome of future infections. Viruses 2019, 11, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paules, C.; Subbarao, K. Influenza. Lancet 2017, 390, 697–708. [Google Scholar] [CrossRef]
- Smith, G.J.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, S.M.; Nunez, A.; Choudhury, B.; Matrosovich, M.; Essen, S.C.; Clifford, D.; Slomka, M.J.; Kuntz-Simon, G.; Garcon, F.; Nash, B.; et al. Replication, pathogenesis and transmission of pandemic (H1N1) 2009 virus in non-immune pigs. PLoS ONE 2010, 5, e9068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Shinya, K.; Kiso, M.; Watanabe, T.; Sakoda, Y.; Hatta, M.; Muramoto, Y.; Tamura, D.; Sakai-Tagawa, Y.; Noda, T.; et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 2009, 460, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Ősterlund, P.; Pirhonen, J.; Ikonen, N.; Rőnkkő, E.; Strengell, M.; Maäkelaä, S.M.; Broman, M.; Hamming, O.J.; Hartmann, R.; Ziegler, T.; et al. Pandemic H1N1 2009 influenza a virus induces weak cytokine responses in human macrophages and dendritic cells and is highly sensitive to the antiviral actions of interferons. J. Virol. 2010, 84, 1414–1422. [Google Scholar] [CrossRef] [Green Version]
- Borgatta, B.; Pérez, M.; Rello, J.; Vidaur, L.; Lorente, L.; Socías, L.; Pozo, J.C.; Pozo, J.; Garnacho-Montero, J.; Rello, J. Elevation of creatine kinase is associated with worse outcomes in 2009 pH1N1 influenza A infection. Intensive Care Med. 2012, 38, 1152–1161. [Google Scholar] [CrossRef]
- Jiang, W.; Liu, S.; Hou, G.; Li, J.; Zhuang, Q.; Wang, S.; Zhang, P.; Chen, J. Chinese and global distribution of H9 subtype avian influenza viruses. PLoS ONE 2012, 7, e52671. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Li, J.; Li, S.; Fu, G.; Jin, T.; Zhang, C.; Yang, Y.; Ma, Z.; Tian, W.; Li, J.; et al. Dominant subtype switch in avian influenza viruses during 2016–2019 in China. Nat. Commun. 2020, 11, 5909. [Google Scholar] [CrossRef]
- Gu, M.; Xu, L.; Wang, X.; Liu, X. Current situation of H9N2 subtype avian influenza in China. Vet. Res. 2017, 48, 49. [Google Scholar] [CrossRef] [Green Version]
- Matrosovich, M.N.; Krauss, S.; Webster, R.G. H9N2 influenza A viruses from poultry in Asia have human virus-like receptor specificity. Virology 2001, 281, 156–162. [Google Scholar] [CrossRef] [Green Version]
- Naguib, M.M.; Arafa, A.S.; Parvin, R.; Beer, M.; Vahlenkamp, T.; Harder, T.C. Insights into genetic diversity and biological propensities of potentially zoonotic avian influenza H9N2 viruses circulating in Egypt. Virology 2017, 511, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Qian, J.; Song, Y.; Ming, D. The adaptability of H9N2 avian influenza A virus to humans: A comparative docking simulation study. Biochem. Biophys. Res. Commun. 2020, 529, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.M.; Li, K.S.; Smith, G.J.; Li, J.W.; Tai, H.; Zhang, J.X.; Webster, R.G.; Peiris, J.S.; Chen, H.; Guan, Y. Evolution and molecular epidemiology of H9N2 influenza A viruses from quail in southern China, 2000 to 2005. J. Virol. 2007, 81, 2635–2645. [Google Scholar] [CrossRef] [Green Version]
- Butt, K.M.; Smith, G.J.; Chen, H.; Zhang, L.J.; Leung, Y.H.; Xu, K.M.; Lim, W.; Webster, R.G.; Yuen, K.Y.; Peiris, J.S.; et al. Human infection with an avian H9N2 influenza A virus in Hong Kong in 2003. J. Clin. Microbiol. 2005, 43, 5760–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, Y.L.; Pu, J.; Liu, Q.F.; Wang, S.; Zhang, G.Z.; Zhang, X.L.; Fan, W.X.; Brown, E.G.; Liu, J.H. Antigenic and genetic characterization of H9N2 swine influenza viruses in China. J. Gen. Virol. 2007, 88 Pt 7, 2035–2041. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, Article17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jozefczuk, J.; Adjaye, J. Quantitative real-time PCR-based analysis of gene expression. Methods Enzym. 2011, 500, 99–109. [Google Scholar]
- Talmi-Frank, D.; Altboum, Z.; Solomonov, I.; Udi, Y.; Jaitin, D.A.; Klepfish, M.; David, E.; Zhuravlev, A.; Keren-Shaul, H.; Winter, D.R.; et al. Extracellular matrix proteolysis by MT1-MMP contributes to influenza-related tissue damage and mortality. Cell Host. Microbe. 2016, 20, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.C.; Chan, R.W.; Wang, J.; Travanty, E.A.; Nicholls, J.M.; Peiris, J.S.; Mason, R.J.; Chan, M.C. Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses. J. Virol. 2011, 85, 6844–6855. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.; Walter, J.M.; Misharin, A.V. Alveolar macrophages. Cell. Immunol. 2018, 330, 86–90. [Google Scholar] [CrossRef]
- Peiró, T.; Patel, D.F.; Akthar, S.; Gregory, L.G.; Pyle, C.J.; Harker, J.A.; Birrell, M.A.; Lloyd, C.M.; Snelgrove, R.J. Neutrophils drive alveolar macrophage IL-1β release during respiratory viral infection. Thorax 2018, 73, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Wang, Q.; Chen, S.; Gao, S.; Song, L.; Liu, P.; Huang, W. Influenza A virus NS1 induces G0/G1 cell cycle arrest by inhibiting the expression and activity of RhoA protein. J. Virol. 2013, 87, 3039–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Zhao, W.; Lu, J.; Li, S.; Zhou, K.; Jiang, W.; Duan, X.; Fu, L.; Yu, B.; Cai, K.Q.; et al. Influenza virus matrix protein M1 interacts with SLD5 to block host cell cycle. Cell Microbiol. 2019, 21, e13038. [Google Scholar] [CrossRef]
- Yang, M.L.; Wang, C.T.; Yang, S.J.; Leu, C.H.; Chen, S.H.; Wu, C.L.; Shiau, A.L. IL-6 ameliorates acute lung injury in influenza virus infection. Sci. Rep. 2017, 7, 43829. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| Apod | GCCACCGACTATGAGAACTATG | CACTGTTTCTGGAGGGAGATTAG |
| IFIT3b | CCTCAGAACCAGTACGTGAAAG | GGAGGACATCCGTTTGATTAG |
| Cmpk2 | CCTGCTCAAACCTGACCTTATC | GGCCTCAAGTTCTGCTTCTT |
| CCL3 | GAAGATTCCACGCCAATTCATC | GATCTGCCGGTTTCTCTTAGTC |
| IL1rn | TTGTGCCAAGTCTGGAGATG | CTCAGAGCGGATGAAGGTAAAG |
| TRIM21 | GATAGCCCAGAATACCAAGAAGAG | GCCCATCTTCCTCACAGAATAG |
| DDX60 | GGTATCCCGATTGGCTGATATG | GAGACACAAGTGGCGAATCT |
| TLR3 | ACCTCCAGAAGAACCTCATAAC | GAACGGATTGAAGCGCATATC |
| CGAS | GGAACCGGACAAGCTAAAGA | CAGGCGTTCCACAACTTTATTC |
| ISG20 | GGAGAGATCACGGACTACAGAA | TAGCCTGGCTTCACCAAATG |
| USP18 | AGAGGACCATGAAGAGGAAGA | CGTCTGTCCGATGTTGTGTAA |
| ACTIN | CCGTAAAGACCTCTATGCCAAC | AGGAGCCAGAGCAGTAATCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, T.; Nagy, A.; Xu, G.; Xin, L.; Bao, D.; Lu, C.; Niu, S.; Wu, Z.; Ren, C.; Zhang, T.; et al. RNA-Seq Analysis of Influenza A Virus-Induced Transcriptional Changes in Mice Lung and Its Possible Implications for the Virus Pathogenicity in Mice. Viruses 2021, 13, 2031. https://doi.org/10.3390/v13102031
Ma T, Nagy A, Xu G, Xin L, Bao D, Lu C, Niu S, Wu Z, Ren C, Zhang T, et al. RNA-Seq Analysis of Influenza A Virus-Induced Transcriptional Changes in Mice Lung and Its Possible Implications for the Virus Pathogenicity in Mice. Viruses. 2021; 13(10):2031. https://doi.org/10.3390/v13102031
Chicago/Turabian StyleMa, Tianxin, Abdou Nagy, Guanlong Xu, Lingxiang Xin, Danqi Bao, Chenyang Lu, Shiqi Niu, Zihua Wu, Chaochao Ren, Ting Zhang, and et al. 2021. "RNA-Seq Analysis of Influenza A Virus-Induced Transcriptional Changes in Mice Lung and Its Possible Implications for the Virus Pathogenicity in Mice" Viruses 13, no. 10: 2031. https://doi.org/10.3390/v13102031
APA StyleMa, T., Nagy, A., Xu, G., Xin, L., Bao, D., Lu, C., Niu, S., Wu, Z., Ren, C., Zhang, T., Yang, J., Teng, Q., Li, X., Li, Z., & Liu, Q. (2021). RNA-Seq Analysis of Influenza A Virus-Induced Transcriptional Changes in Mice Lung and Its Possible Implications for the Virus Pathogenicity in Mice. Viruses, 13(10), 2031. https://doi.org/10.3390/v13102031