
Genome Characterization of Bird-Related Rhabdoviruses Circulating in Africa


 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Description
2.2. RNA Extraction
2.3. Genome Sequence Determination
2.4. Sequences Analysis
3. Results
3.1. Genome Characterization of the Bird-Related Rhabdoviruses
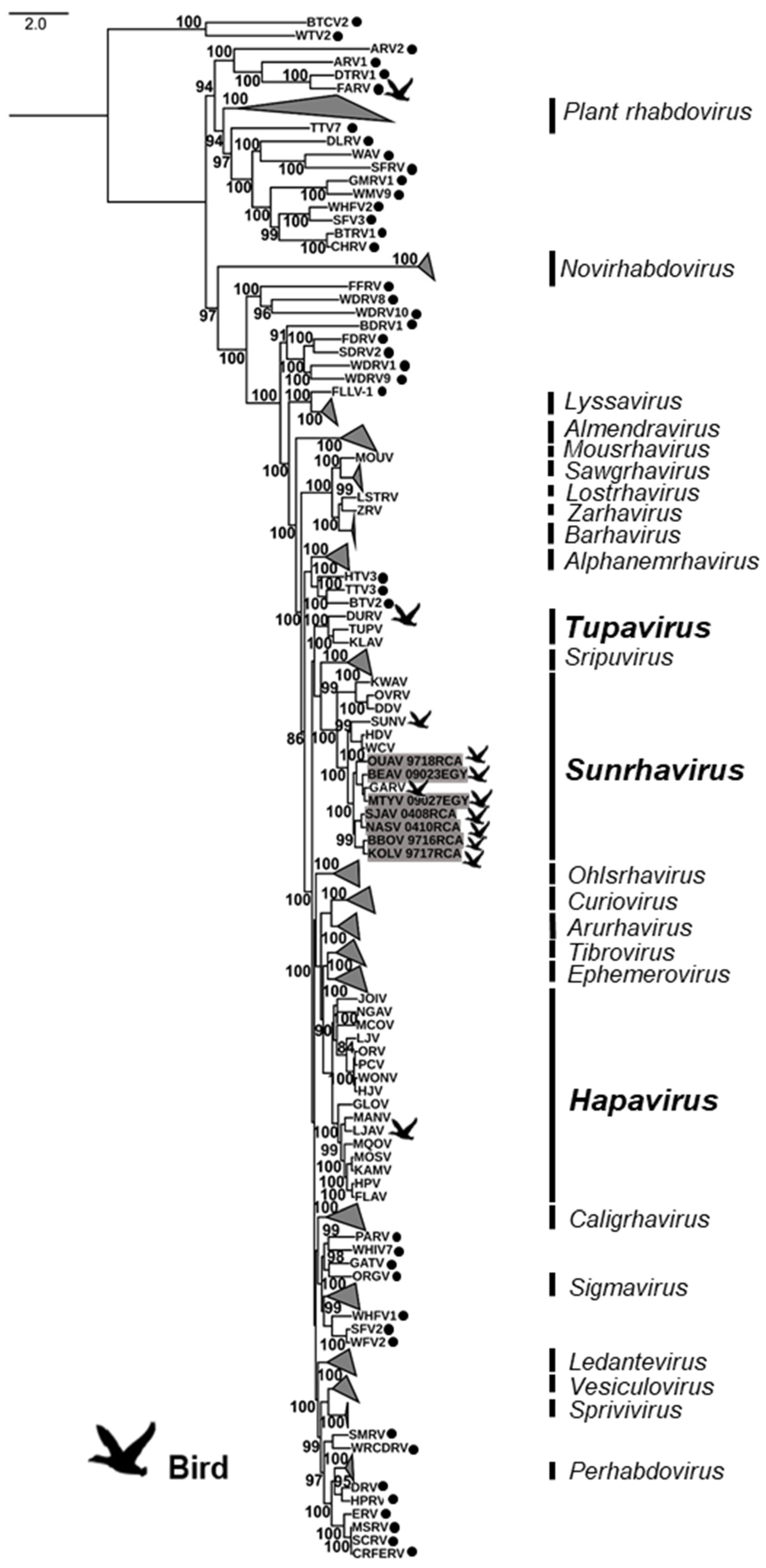
3.2. Phylogenetic Analysis of the Bird-Related Rhabdoviruses
3.3. Genetic Diversity of the Bird-Related Rhabdoviruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, P.J.; Firth, C.; Widen, S.G.; Blasdell, K.R.; Guzman, H.; Wood, T.G.; Paradkar, P.N.; Holmes, E.C.; Tesh, R.B.; Vasilakis, N. Evolution of Genome Size and Complexity in the Rhabdoviridae. PLoS Pathog. 2015, 11, e1004664. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, J.H.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Amarasinghe, G.K.; Anthony, S.J.; Avšič-Županc, T.; Ayllón, M.A.; Bahl, J.; Balkema-Buschmann, A.; et al. 2020 Taxonomic Update for Phylum Negarnaviricota (Riboviria: Orthornavirae), Including the Large Orders Bunyavirales and Mononegavirales. Arch. Virol. 2020, 50, 3023–3072. [Google Scholar] [CrossRef]
- Walker, P.J.; Dietzgen, R.G.; Joubert, D.A.; Blasdell, K.R. Rhabdovirus Accessory Genes. Virus Res. 2011, 162, 110–125. [Google Scholar] [CrossRef]
- Walker, P.J.; Blasdell, K.R.; Calisher, C.H.; Dietzgen, R.G.; Kondo, H.; Kurath, G.; Longdon, B.; Stone, D.M.; Tesh, R.B.; Tordo, N.; et al. ICTV Virus Taxonomy Profile: Rhabdoviridae. J. Gen. Virol. 2018, 99, 447–448. [Google Scholar] [CrossRef] [PubMed]
- Calisher, C.H.; Karabatsos, N.; Zeller, H.; Digoutte, J.-P.; Tesh, R.B.; Shope, R.E.; da Rosa, A.P.A.T.; George, T.D.S. Antigenic Relationships among Rhabdoviruses from Vertebrates and Hematophagous Arthropods. Intervirology 1989, 30, 241–257. [Google Scholar] [CrossRef]
- Clark, G.G.; Taylor, D.E.; Crabbs, C.L.; Calisher, C.H.; Bowen, R.A.; Tesh, R.B.; Canestorp, K.M. Malpais Spring Virus: A New Vesiculovirus from Mosquitoes Collected in New Mexico and Evidence of Infected Indigenous and Exotic Ungulates. Am. J. Trop. Med. Hyg. 1988, 39, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Kurz, W.; Gelderblom, H.; Flügel, R.M.; Darai, G. Isolation and Characterization of a Tupaia Rhabdovirus. Intervirology 1986, 25, 88–96. [Google Scholar] [CrossRef]
- Bourhy, H.; Sureau, P.; Tordo, N. From Rabies to Rabies-Related Viruses. Vet. Microbiol. 1990, 23, 115–128. [Google Scholar] [CrossRef]
- Gibbs, A.J. Viral Taxonomy Needs a Spring Clean; Its Exploration Era Is Over. Virol. J. 2013, 10, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- Allison, A.B.; Palacios, G.; Travassos da Rosa, A.; Popov, V.L.; Lu, L.; Xiao, S.Y.; DeToy, K.; Briese, T.; Lipkin, W.I.; Keel, M.K.; et al. Characterization of Durham Virus, a Novel Rhabdovirus That Encodes Both a C and SH Protein. Virus Res. 2011, 155, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.P.; Zeidner, N.; Borland, E.M.; Mutebi, J.-P.; Lanciotti, R.S.; Miller, B.R.; Lutwama, J.J.; Tendo, J.M.; Andama, V.; Powers, A.M. Sunguru Virus: A Novel Virus in the Family Rhabdoviridae Isolated from a Chicken in North-Western Uganda. J. Gen. Virol. 2014, 95, 1436–1443. [Google Scholar] [CrossRef]
- Palacios, G.; Forrester, N.L.; Savji, N.; Travassos da Rosa, A.P.A.; Guzman, H.; DeToy, K.; Popov, V.L.; Walker, P.J.; Lipkin, W.; Vasilakis, N.; et al. Characterization of Farmington Virus, a Novel Virus from Birds That Is Distantly Related to Members of the Family Rhabdoviridae. Virol. J. 2013, 10, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Haas, R.A.; Jonkers, A.H.; Heinemann, D.W. Kwatta Virus, a New Agent Isolated from Culex Mosquitoes in Surinam *. Am. J. Trop. Med. Hyg. 1966, 15, 954–957. [Google Scholar] [CrossRef]
- Mekki, A.A.E.; Nieuwenhuysen, P.; van der Groen, G.; Pattyn, S.R. Characterization of Some Ungrouped Viruses. Trans. R. Soc. Trop. Med. Hyg. 1981, 75, 799–806. [Google Scholar] [CrossRef]
- Dacheux, L.; Berthet, N.; Dissard, G.; Holmes, E.C.; Delmas, O.; Larrous, F.; Guigon, G.; Dickinson, P.; Faye, O.; Sall, A.A.; et al. Application of Broad-Spectrum Resequencing Microarray for Genotyping Rhabdoviruses. J. Virol. 2010, 84, 9557–9574. [Google Scholar] [CrossRef] [Green Version]
- McAllister, J.; Gauci, P.J.; Mitchell, I.R.; Boyle, D.B.; Bulach, D.M.; Weir, R.P.; Melville, L.F.; Davis, S.S.; Gubala, A.J. Genomic Characterisation of Almpiwar Virus, Harrison Dam Virus and Walkabout Creek Virus; Three Novel Rhabdoviruses from Northern Australia. Virol. Rep. 2014, 3–4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kuzmin, I.V. Phylogenetic Relationships of Seven Previously Unclassified Viruses within the Family Rhabdoviridae Using Partial Nucleoprotein Gene Sequences. J. Gen. Virol. 2006, 87, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Quan, P.-L.; Williams, D.T.; Johansen, C.A.; Jain, K.; Petrosov, A.; Diviney, S.M.; Tashmukhamedova, A.; Hutchison, S.K.; Tesh, R.B.; Mackenzie, J.S.; et al. Genetic Characterization of K13965, a Strain of Oak Vale Virus from Western Australia. Virus Res. 2011, 160, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Tesh, R.B.; Travassos Da Rosa, A.P.A.; Travassos Da Rosa, J.S.Y. Antigenic Relationship Among Rhabdoviruses Infecting Terrestrial Vertebrates. J. Gen. Virol. 1983, 64, 169–176. [Google Scholar] [CrossRef]
- Pallandre, L.; Luo, D.; Feuvrier, C.; Lieffrig, F.; Pozet, F.; Dacheux, L.; Bigarré, L. Revisiting the Classification of Percid Perhabdoviruses Using New Full-Length Genomes. Viruses 2020, 12, 649. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.-S.; Li, B.; Shen, X.-R.; Jiang, R.-D.; Zhu, Y.; Wu, J.; Fan, Y.; Bourhy, H.; Hu, B.; Ge, X.-Y.; et al. Characterization of Novel Rhabdoviruses in Chinese Bats. Viruses 2021, 13, 64. [Google Scholar] [CrossRef]
- Dacheux, L.; Dommergues, L.; Chouanibou, Y.; Doméon, L.; Schuler, C.; Bonas, S.; Luo, D.; Maufrais, C.; Cetre-Sossah, C.; Cardinale, E.; et al. Co-circulation and Characterization of Novel African Arboviruses (Genus Ephemerovirus) in Cattle, Mayotte Island, Indian Ocean, 2017. Transbound. Emerg. Dis. 2019, 66, 2601–2604. [Google Scholar] [CrossRef] [Green Version]
- Mareuil, F.; Doppelt-Azeroual, O.; Ménager, H. A Public Galaxy Platform at Pasteur Used as an Execution Engine for Web Services. F1000Research 2017, 6, 1030. [Google Scholar] [CrossRef]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for Visual Exploration of Second-Generation Sequencing Data. Brief. Bioinform. 2012, 14, 193–202. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Kall, L.; Krogh, A.; Sonnhammer, E.L.L. Advantages of Combined Transmembrane Topology and Signal Peptide Prediction--the Phobius Web Server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [Green Version]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to Virus Taxonomy and to the International Code of Virus Classification and Nomenclature Ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef] [PubMed]
- Peluso, R.W.; Richardson, J.C.; Talon, J.; Lock, M. Identification of a Set of Proteins (C′ and C) Encoded by the Bicistronic P Gene of the Indiana Serotype of Vesicular Stomatitis Virus and Analysis of Their Effect on Transcription by the Viral RNA Polymerase. Virology 1996, 218, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Nadin-Davis, S.A.; Abdel-Malik, M.; Armstrong, J.; Wandeler, A.I. Lyssavirus P Gene Characterisation Provides Insights into the Phylogeny of the Genus and Identifies Structural Similarities and Diversity within the Encoded Phosphoprotein. Virology 2002, 298, 286–305. [Google Scholar] [CrossRef] [Green Version]
- Walker, P.J. Bovine Ephemeral Fever in Australia and the World. In The World of Rhabdoviruses; Fu, Z.F., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 292, pp. 57–80. [Google Scholar]
- Reeves, W.K.; Miller, M.M.; Gruner, W.E. Two Rhabdoviridae: Dillard’s Draw Virus, a Putative New Virus, and Merida Virus from Culex Tarsalis (Diptera: Culicidae) in New Mexico, USA. Acta Virol. 2018, 62, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Springfeld, C.; Darai, G.; Cattaneo, R. Characterization of the Tupaia Rhabdovirus Genome Reveals a Long Open Reading Frame Overlapping with P and a Novel Gene Encoding a Small Hydrophobic Protein. J. Virol. 2005, 79, 6781–6790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montecino-Latorre, D.; Barker, C.M. Overwintering of West Nile Virus in a Bird Community with a Communal Crow Roost. Sci. Rep. 2018, 8, 6088. [Google Scholar] [CrossRef] [Green Version]
- Vilibic-Cavlek, T.; Petrovic, T.; Savic, V.; Barbic, L.; Tabain, I.; Stevanovic, V.; Klobucar, A.; Mrzljak, A.; Ilic, M.; Bogdanic, M.; et al. Epidemiology of Usutu Virus: The European Scenario. Pathogens 2020, 9, 699. [Google Scholar] [CrossRef]
- Farajollahi, A.; Fonseca, D.M.; Kramer, L.D.; Marm Kilpatrick, A. “Bird Biting” Mosquitoes and Human Disease: A Review of the Role of Culex Pipiens Complex Mosquitoes in Epidemiology. Infect. Genet. Evol. 2011, 11, 1577–1585. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Isolate | Name | Acronym | City | Country | Date of Collection | Host Species | Original Samples | Tested Samples |
|---|---|---|---|---|---|---|---|---|
| 9716RCA | Bimbo virus | BBOV | Kolongo | Central African Republic | 18 July 1970 | Euplectes afra | Unknown | Mouse brain |
| 9717RCA | Kolongo virus | KOLV | Bangui | Central African Republic | 18 July 1970 | Euplectes afra | Pool of crushed organs | Mouse brain |
| 9718RCA | Ouango virus | OUAV | Landjia | Central African Republic | 10 October 1970 | Ploceus melanocephalus | Blood | Mouse brain |
| 0408RCA | Sandjimba virus | SJAV | Landjia | Central African Republic | 21 January 1970 | Acrocephalus schoenbaeus | Pool of crushed organs | Mouse brain |
| 0410RCA | Nasoule virus | NASV | Nasoulé | Central African Republic | 15 September 1973 | Andropadus virens | Blood | Mouse brain |
| 09023EGY | Burg el Arab virus | BEAV | Bahig | Egypt | 15 October 1962 | Sylvia curraca | Unknown | Mouse brain |
| 09027EGY | Matariya virus | MTYV | Port Saïd | Egypt | 8 October 1961 | Sylvia curraca | Unknown | Mouse brain |
| Isolate | Virus | Genome Size (nt) | Raw Reads (no) | Reads Cleaned (no) | Mapped Reads (no) | Average Coverage Depth * (x) | GenBank Accession Number |
|---|---|---|---|---|---|---|---|
| 9716RCA | BBOV | 10,969 | 3,547,036 | 3,013,558 | 15,226 | 206.23 | MW491756 |
| 9717RCA | KOLV | 10,971 | 4,881,362 | 4,214,012 | 46,475 | 629.49 | MW491757 |
| 9718RCA | OUAV | 10,805 | 12,256,546 | 9,754,242 | 28,150 | 384.67 | MW491758 |
| 0408RCA | SJAV | 10,951 | 12,164,616 | 9,908,488 | 14,166 | 191.80 | MW491754 |
| 0410RCA | NASV | 10,977 | 8,689,566 | 6,630,310 | 20,937 | 281.01 | MW491755 |
| 09023EGY | BEAV | 10,846 | 101,186 | 65,258 | 832 | 11.15 | MW491759 |
| 09027EGY | MTYV | 11,021 | 2,742,712 | 2,289,168 | 3635 | 45.58 | MW491760 |
| SJAV 0408RCA | NASV 0410RCA | BBOV 9716RCA | KOLV 9717RCA | OUAV 9718RCA | BEAV 09023EGY | MTYV 09027EGY | GARV | HDV | WCV | SUNV | DDV | OVRV | KWAV | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SJAV 0408RCA | ||||||||||||||
| NASV 0410RCA | 71.9 | |||||||||||||
| BBOV 9716RCA | 57.9 | 56.7 | ||||||||||||
| KOLV 9717RCA | 57.9 | 56.2 | 71.3 | |||||||||||
| OUAV 9718RCA | 58.4 | 56.8 | 56 | 55.7 | ||||||||||
| BEAV 09023EGY | 57.8 | 56.3 | 56.1 | 55.5 | 58.4 | |||||||||
| MTYV 09027EGY | 58.2 | 56.7 | 56.4 | 55.8 | 58.8 | 70.1 | ||||||||
| GARV | 58.2 | 56.5 | 55.5 | 55.7 | 59 | 69.2 | 73.2 | |||||||
| HDV | 51.7 | 50 | 50.8 | 50.5 | 51.5 | 51.5 | 51 | 51.1 | ||||||
| WCV | 50.6 | 48.9 | 49.8 | 49.4 | 50.6 | 51.3 | 50.6 | 50.5 | 74.8 | |||||
| SUNV | 48.9 | 48.1 | 47.5 | 47.2 | 48.6 | 48.8 | 47.9 | 48.2 | 54 | 53.8 | ||||
| DDV | 42.4 | 41.6 | 42.3 | 41.8 | 42.4 | 42.4 | 42.3 | 42.1 | 43.4 | 42.5 | 41.5 | |||
| OVRV | 41 | 40.5 | 41 | 41.5 | 41 | 41.8 | 41.6 | 41.4 | 42.4 | 41.9 | 40.8 | 65.2 | ||
| KWAV | 42.3 | 41.4 | 41.9 | 41.7 | 41.9 | 41.6 | 41.8 | 42 | 42.7 | 42.1 | 41.2 | 52.1 | 51.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, D.-S.; Zhou, Z.-J.; Ge, X.-Y.; Bourhy, H.; Shi, Z.-L.; Grandadam, M.; Dacheux, L. Genome Characterization of Bird-Related Rhabdoviruses Circulating in Africa. Viruses 2021, 13, 2168. https://doi.org/10.3390/v13112168
Luo D-S, Zhou Z-J, Ge X-Y, Bourhy H, Shi Z-L, Grandadam M, Dacheux L. Genome Characterization of Bird-Related Rhabdoviruses Circulating in Africa. Viruses. 2021; 13(11):2168. https://doi.org/10.3390/v13112168
Chicago/Turabian StyleLuo, Dong-Sheng, Zhi-Jian Zhou, Xing-Yi Ge, Hervé Bourhy, Zheng-Li Shi, Marc Grandadam, and Laurent Dacheux. 2021. "Genome Characterization of Bird-Related Rhabdoviruses Circulating in Africa" Viruses 13, no. 11: 2168. https://doi.org/10.3390/v13112168
APA StyleLuo, D. -S., Zhou, Z. -J., Ge, X. -Y., Bourhy, H., Shi, Z. -L., Grandadam, M., & Dacheux, L. (2021). Genome Characterization of Bird-Related Rhabdoviruses Circulating in Africa. Viruses, 13(11), 2168. https://doi.org/10.3390/v13112168