
Phylogenetic Characteristics of Canine Parvovirus Type 2c Variant Endemic in Shanghai, China

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Viral DNA Extraction and Amplification
2.3. Virus Isolation
2.4. Immunofluorescence Assay
2.5. Near Full-Length Genome Sequencing of CPV-2 Isolates
2.6. Phylogenetic Analysis
2.7. Structural Analysis of VP2 Protein
2.8. Selection Pressure Site Analysis
3. Results
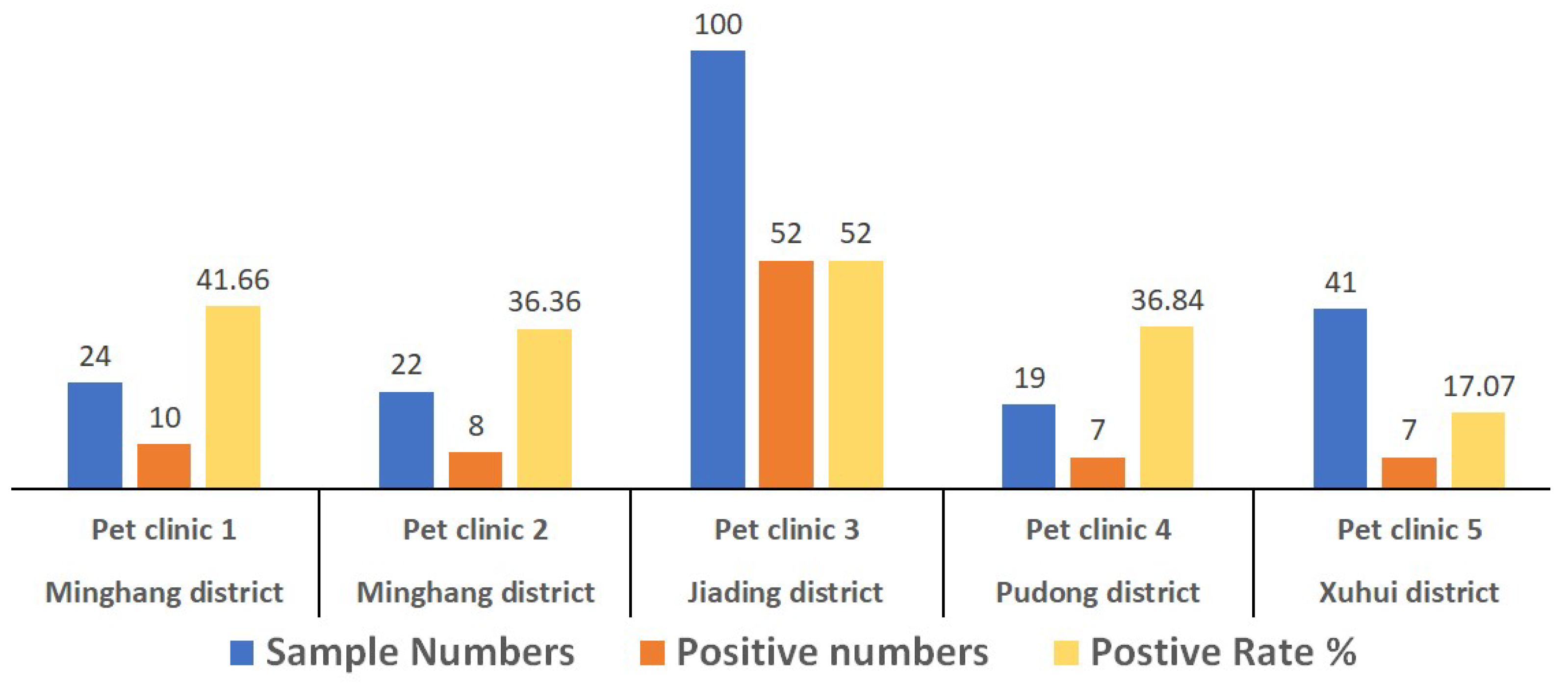
3.1. Positive Detection Rate of CPV-2 in Canine Patients
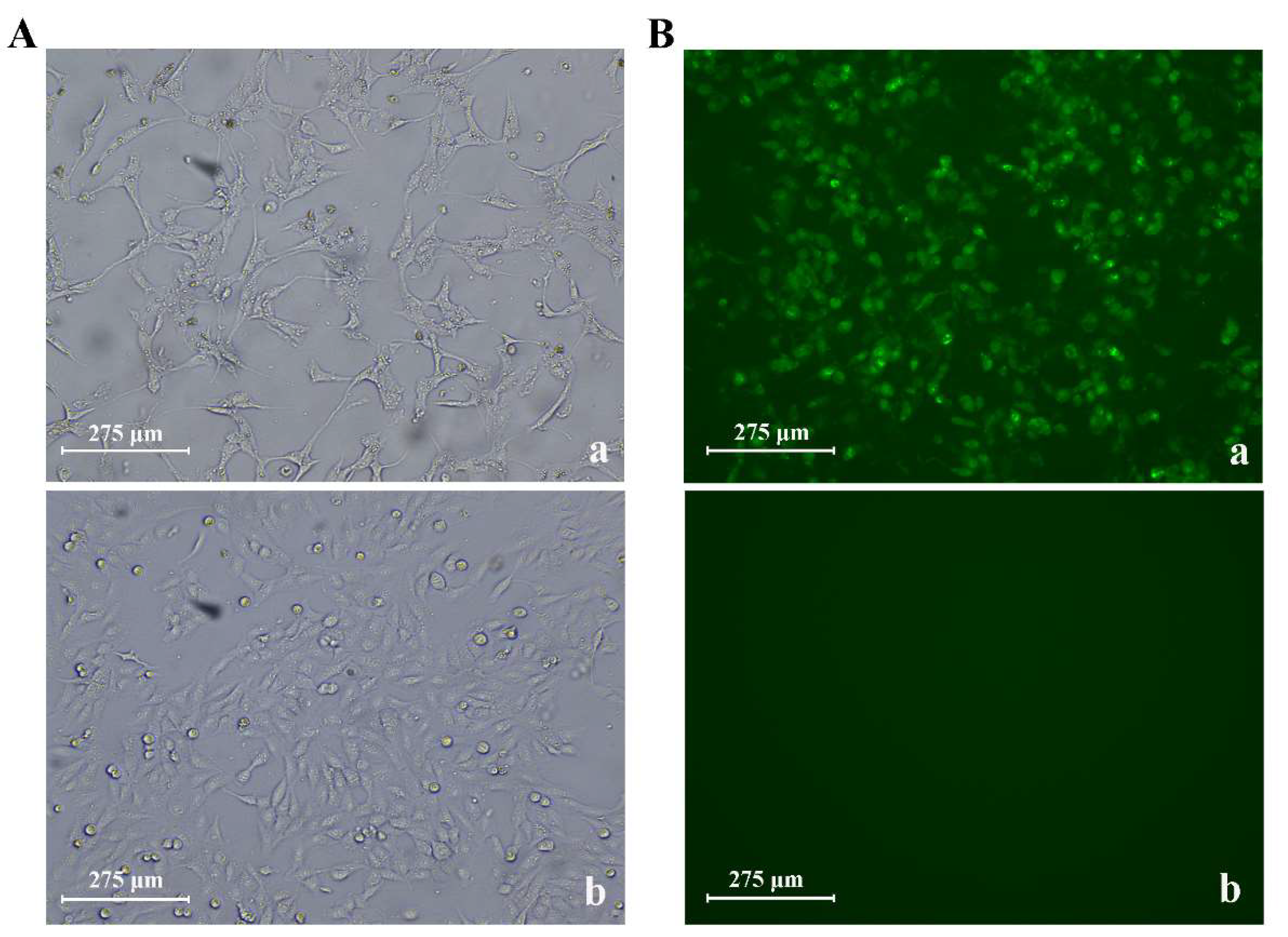
3.2. CPV Isolation and Identification
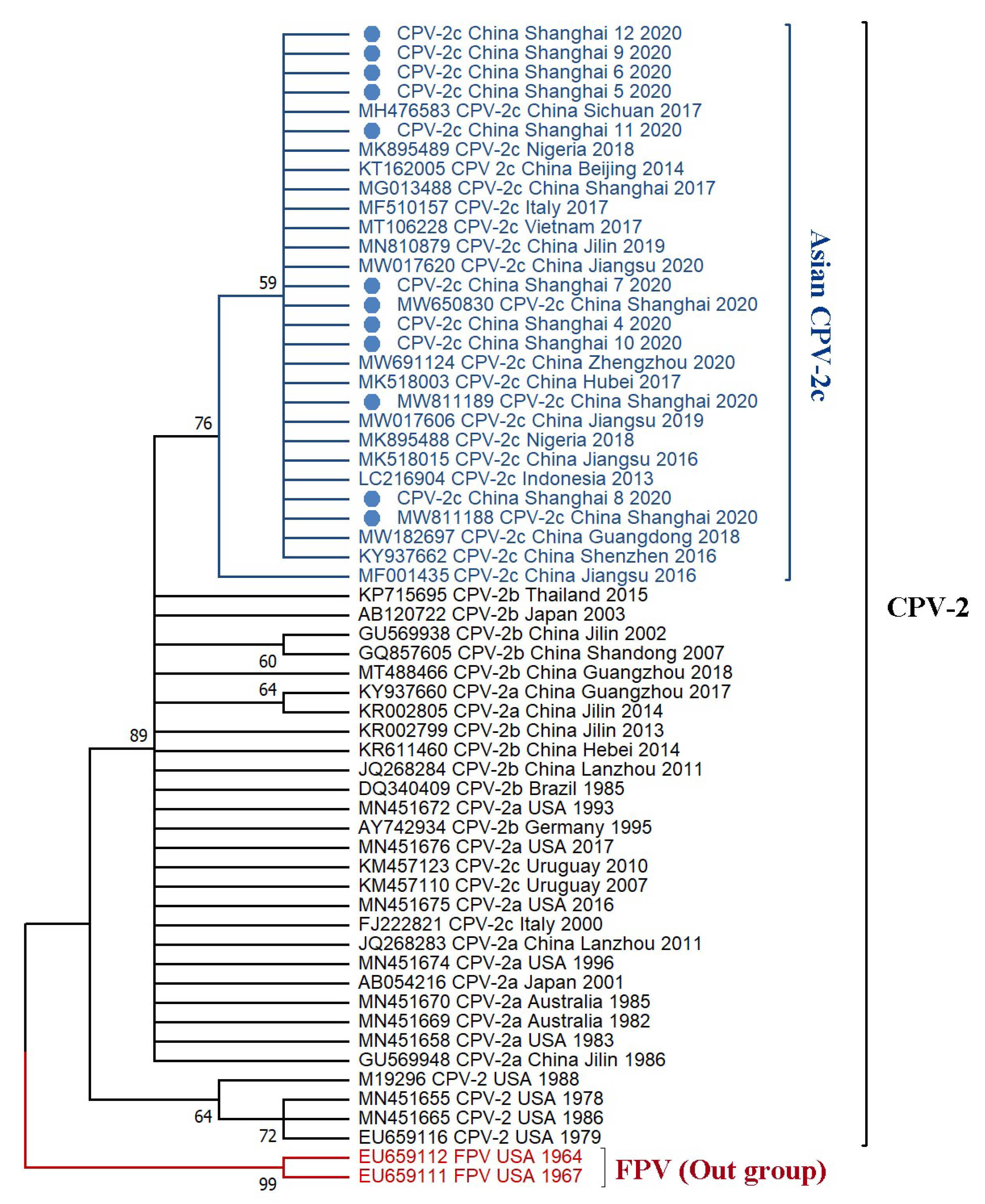
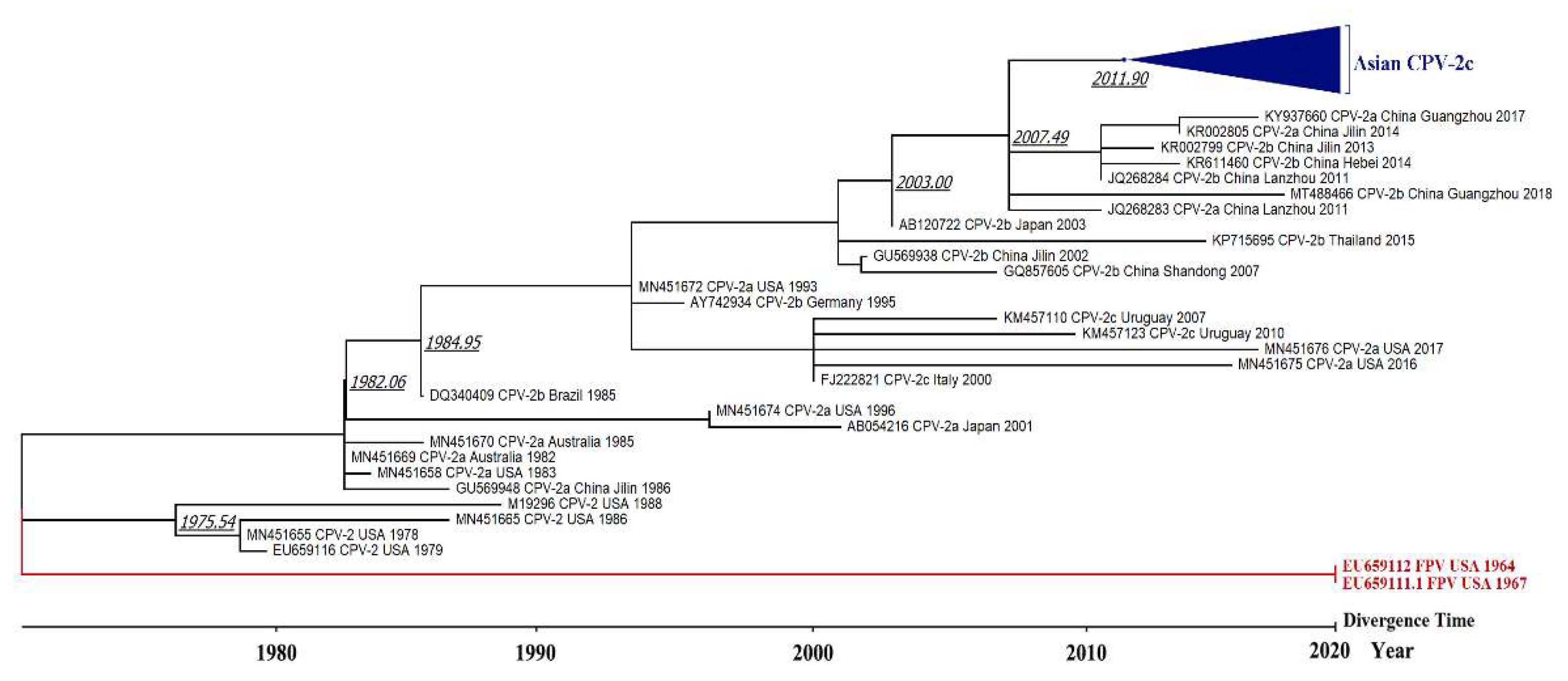
3.3. Phylogenetic Tree and Divergence Time Estimate
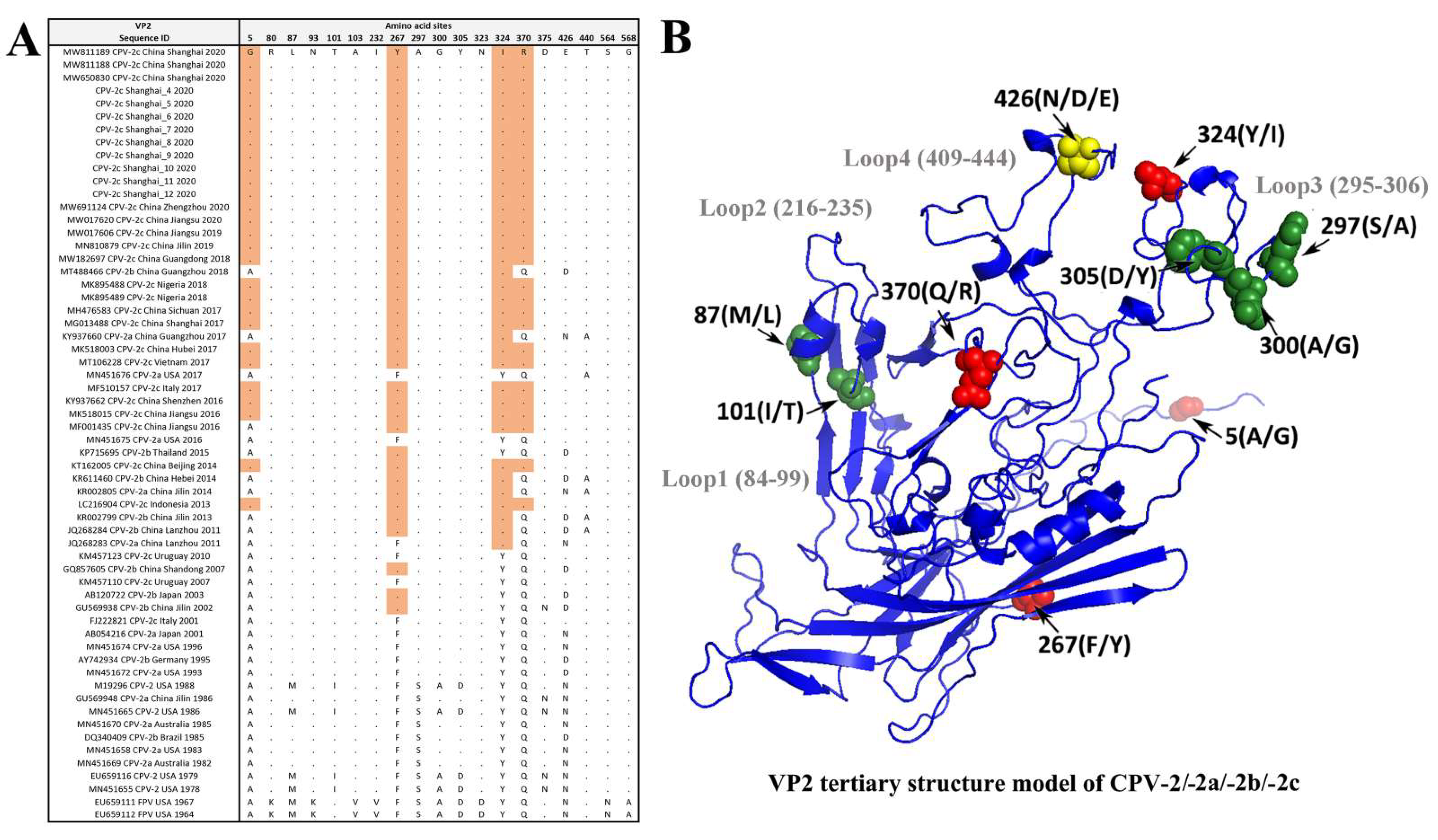
3.4. Amino Acid Site Analysis of the VP2 Gene
3.5. Positive Selection Pressure Sites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Appel, M.; Scott, F.; Carmichael, L. Isolation and immunisation studies of a canine parco-like virus from dogs with haemorrhagic enteritis. Vet. Rec. 1979, 105, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Burtonboy, G.; Coignoul, F.; Delferriere, N.; Pastoret, P.-P. Canine hemorrhagic enteritis: Detection of viral particles by electron microscopy. Arch. Virol. 1979, 61, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, I.E.; Lee, H.; Allison, A.B.; Lopez-Astacio, R.; Goodman, L.B.; Oyesola, O.O.; Omobowale, O.; Fagbohun, O.; Dubovi, E.J.; Hafenstein, S.L. Limited intrahost diversity and background evolution accompany 40 years of canine parvovirus host adaptation and spread. J. Virol. 2019, 94, e01162-19. [Google Scholar] [CrossRef]
- Hueffer, K.; Parker, J.S.; Weichert, W.S.; Geisel, R.E.; Sgro, J.-Y.; Parrish, C.R. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J. Virol. 2003, 77, 1718–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truyen, U.; Gruenberg, A.; Chang, S.-F.; Obermaier, B.; Veijalainen, P.; Parrish, C.R. Evolution of the feline-subgroup parvoviruses and the control of canine host range in vivo. J. Virol. 1995, 69, 4702–4710. [Google Scholar] [CrossRef] [Green Version]
- Reed, A.P.; Jones, E.V.; Miller, T.J. Nucleotide sequence and genome organization of canine parvovirus. J. Virol. 1988, 62, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Zeng, W.; Zhang, X.; Li, S. The genetic evolution of canine parvovirusA new perspective. PLoS ONE 2017, 12, e0175035. [Google Scholar]
- Binn, L.; Lazar, E.; Eddy, G.; Kajima, M. Recovery and characterization of a minute virus of canines. Infect. Immun. 1970, 1, 503–508. [Google Scholar] [CrossRef] [Green Version]
- Decaro, N.; Amorisco, F.; Lenoci, D.; Lovero, A.; Colaianni, M.L.; Losurdo, M.; Desario, C.; Martella, V.; Buonavoglia, C. Molecular characterization of Canine minute virus associated with neonatal mortality in a litter of Jack Russell terrier dogs. J. Vet. Diagn. Investig. 2012, 24, 755–758. [Google Scholar] [CrossRef] [Green Version]
- Parrish, C.R.; Have, P.; Foreyt, W.J.; Evermann, J.F.; Senda, M.; Carmichael, L.E. The global spread and replacement of canine parvovirus strains. J. Gen. Virol. 1988, 69, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Aquadro, C.F.; Strassheim, M.; Evermann, J.; Sgro, J.; Mohammed, H. Rapid antigenic-type replacement and DNA sequence evolution of canine parvovirus. J. Virol. 1991, 65, 6544–6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonavoglia, C.; Martella, V.; Pratelli, A.; Tempesta, M.; Cavalli, A.; Buonavoglia, D.; Bozzo, G.; Elia, G.; Decaro, N.; Carmichael, L. Evidence for evolution of canine parvovirus type 2 in Italy. J. Gen. Virol. 2001, 82, 3021–3025. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; He, Y.; Wang, C.; Xiao, W.; Liu, R.; Xiao, X.; Zhou, P.; Li, S. The increasing prevalence of CPV-2c in domestic dogs in China. Peer J. 2020, 8, e9869. [Google Scholar] [CrossRef] [PubMed]
- Temuujin, U.; Tserendorj, A.; Fujiki, J.; Sakoda, Y.; Tseren-Ochir, E.-O.; Okamatsu, M.; Matsuno, K.; Sharav, T.; Horiuchi, M.; Umemura, T. The first isolation and identification of canine parvovirus (CPV) type 2c variants during 2016–2018 genetic surveillance of dogs in Mongolia. Infect. Genet. Evol. 2019, 73, 269–275. [Google Scholar] [CrossRef]
- Hoang, M.; Lin, W.-H.; Nga, B.T.T.; Chiou, M.-T.; Lin, C.-N. Molecular epidemiology of canine parvovirus type 2 in Vietnam from November 2016 to February 2018. Virol. J. 2019, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Balboni, A.; Niculae, M.; Di Vito, S.; Urbani, L.; Terrusi, A.; Muresan, C.; Battilani, M.J.B.v.r. The detection of canine parvovirus type 2c of Asian origin in dogs in Romania evidenced its progressive worldwide diffusion. BMC Veter Res. 2021, 17, 1–6. [Google Scholar]
- Zhao, H.; Wang, J.; Jiang, Y.; Cheng, Y.; Lin, P.; Zhu, H.; Han, G.; Yi, L.; Zhang, S.; Guo, L.J.T.; et al. Typing of canine parvovirus strains circulating in North-East China. Transbound Emerg. Dis. 2017, 64, 495–503. [Google Scholar] [CrossRef]
- Wang, J.; Lin, P.; Zhao, H.; Cheng, Y.; Jiang, Z.; Zhu, H.; Wu, H.; Cheng, S.J.I.; Genetics; Evolution. Continuing evolution of canine parvovirus in China: Isolation of novel variants with an Ala5Gly mutation in the VP2 protein. Infect. Genet. Evol. 2016, 38, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, J.; Bi, Z.; Tan, Y.; Lv, L.; Zhao, H.; Xia, X.; Zhu, Y.; Wang, Y.; Qian, J.J.I.; et al. Molecular epidemiology and genetic evolution of canine parvovirus in East China, during 2018–2020. Infect. Genet. Evol. 2021, 90, 104780. [Google Scholar] [CrossRef]
- Mira, F.; Purpari, G.; Lorusso, E.; Di Bella, S.; Gucciardi, F.; Desario, C.; Macaluso, G.; Decaro, N.; Guercio, A.J.T. Introduction of Asian canine parvovirus in Europe through dog importation. Transbound. Emerg. Dis. 2018, 65, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Mira, F.; Purpari, G.; Di Bella, S.; Colaianni, M.L.; Schirò, G.; Chiaramonte, G.; Gucciardi, F.; Pisano, P.; Lastra, A.; Decaro, N.J.T.; et al. Spreading of canine parvovirus type 2c mutants of Asian origin in southern Italy. Transbound. Emerg. Dis. 2019, 66, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Ogbu, K.I.; Mira, F.; Purpari, G.; Nwosuh, C.; Loria, G.R.; Schirò, G.; Chiaramonte, G.; Tion, M.T.; Di Bella, S.; Ventriglia, G.J.T.; et al. Nearly full-length genome characterization of canine parvovirus strains circulating in Nigeria. Transbound. Emerg. Dis. 2020, 67, 635–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Tsao, J.; Chapman, M.S.; Agbandje, M.; Keller, W.; Smith, K.; Wu, H.; Luo, M.; Smith, T.J.; Rossmann, M.G.; Compans, R.W. The three-dimensional structure of canine parvovirus and its functional implications. Science 1991, 251, 1456–1464. [Google Scholar] [CrossRef] [Green Version]
- Simpson, A.A.; Chandrasekar, V.; Hébert, B.T.; Sullivan, G.M.; Rossmann, M.G.; Parrish, C.R. Host range and variability of calcium binding by surface loops in the capsids of canine and feline parvoviruses. J. Mol. Biol. 2000, 300, 597–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L. The PyMOL molecular graphics system, version 1.8. 2015. [Google Scholar]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.F.; Sgro, J.Y.; Parrish, C.R. Multiple amino acids in the capsid structure of canine parvovirus coordinately determine the canine host range and specific antigenic and hemagglutination properties. J. Virol. 1992, 66, 6858–6867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truyen, U.; Platzer, G.; Parrish, C. Antigenic type distribution among canine parvoviruses in dogs and cats in Germany. Vet. Rec. 1996, 138, 365. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.B.; Organtini, L.J.; Zhang, S.; Hafenstein, S.L.; Holmes, E.C.; Parrish, C.R. Single mutations in the VP2 300 loop region of the three-fold spike of the carnivore parvovirus capsid can determine host range. J. Virol. 2016, 90, 753–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraldo-Ramirez, S.; Rendon-Marin, S.; Ruiz-Saenz, J. Phylogenetic, evolutionary and structural analysis of Canine Parvovirus (CPV-2) antigenic variants circulating in Colombia. Viruses 2020, 12, 500. [Google Scholar] [CrossRef]
- Siedek, E.M.; Schmidt, H.; Sture, G.H.; Raue, R. Vaccination with canine parvovirus type 2 (CPV-2) protects against challenge with virulent CPV-2b and CPV-2c. Berl. und Munch. Tierarztl. Wochenschr. 2011, 124, 58–64. [Google Scholar]
- Decaro, N.; Desario, C.; Elia, G.; Martella, V.; Mari, V.; Lavazza, A.; Nardi, M.; Buonavoglia, C. Evidence for immunisation failure in vaccinated adult dogs infected with canine parvovirus type 2c. Microbiol. Q. J. Microbiol. Sci. 2008, 31, 125–130. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | α | β | β-α | Prob [α > β] | Prob [α < β] | BayesFactor [α < β] |
|---|---|---|---|---|---|---|
| 426 | 3.971 | 46.531 | 42.560 | 0.005 | 0.966 1 | 45.222 |
| 324 | 3.948 | 35.271 | 31.323 | 0.024 | 0.943 1 | 26.092 |
| 375 | 4.632 | 23.224 | 18.591 | 0.066 | 0.892 | 13.063 |
| 440 | 2.471 | 14.691 | 12.219 | 0.079 | 0.885 | 12.114 |
| 13 | 2.946 | 15.705 | 12.759 | 0.084 | 0.878 | 11.339 |
| 267 | 4.632 | 14.76 | 10.128 | 0.126 | 0.828 | 7.613 |
| 87 | 1.072 | 5.364 | 4.293 | 0.222 | 0.733 | 4.332 |
| 370 | 3.086 | 6.645 | 3.559 | 0.267 | 0.684 | 3.419 |
| 579 | 2.298 | 3.554 | 1.255 | 0.296 | 0.652 | 2.969 |
| 5 | 2.297 | 3.484 | 1.187 | 0.297 | 0.651 | 2.949 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Gao, J.; Li, H.; Sun, F.; Liang, H.; Liu, H.; Yi, J. Phylogenetic Characteristics of Canine Parvovirus Type 2c Variant Endemic in Shanghai, China. Viruses 2021, 13, 2257. https://doi.org/10.3390/v13112257
Liu C, Gao J, Li H, Sun F, Liang H, Liu H, Yi J. Phylogenetic Characteristics of Canine Parvovirus Type 2c Variant Endemic in Shanghai, China. Viruses. 2021; 13(11):2257. https://doi.org/10.3390/v13112257
Chicago/Turabian StyleLiu, Chengqian, Jun Gao, Hong Li, Fengping Sun, Hongyu Liang, Huili Liu, and Jianzhong Yi. 2021. "Phylogenetic Characteristics of Canine Parvovirus Type 2c Variant Endemic in Shanghai, China" Viruses 13, no. 11: 2257. https://doi.org/10.3390/v13112257
APA StyleLiu, C., Gao, J., Li, H., Sun, F., Liang, H., Liu, H., & Yi, J. (2021). Phylogenetic Characteristics of Canine Parvovirus Type 2c Variant Endemic in Shanghai, China. Viruses, 13(11), 2257. https://doi.org/10.3390/v13112257