
Analyses of Leishmania-LRV Co-Phylogenetic Patterns and Evolutionary Variability of Viral Proteins

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Leishmania spp. Strains, Cultivation, and Screening for dsRNA Viruses
2.2. Whole-Genome Sequencing and Read Mapping
2.3. Phylogenetic Analyses of Leishmania major Strains
2.4. Phylogenetic Analyses of LRVs
2.5. Structure Prediction and Functional Sites Annotation
3. Results
3.1. Screening of Leishmania spp. Isolates for the Presence of dsRNA Viruses
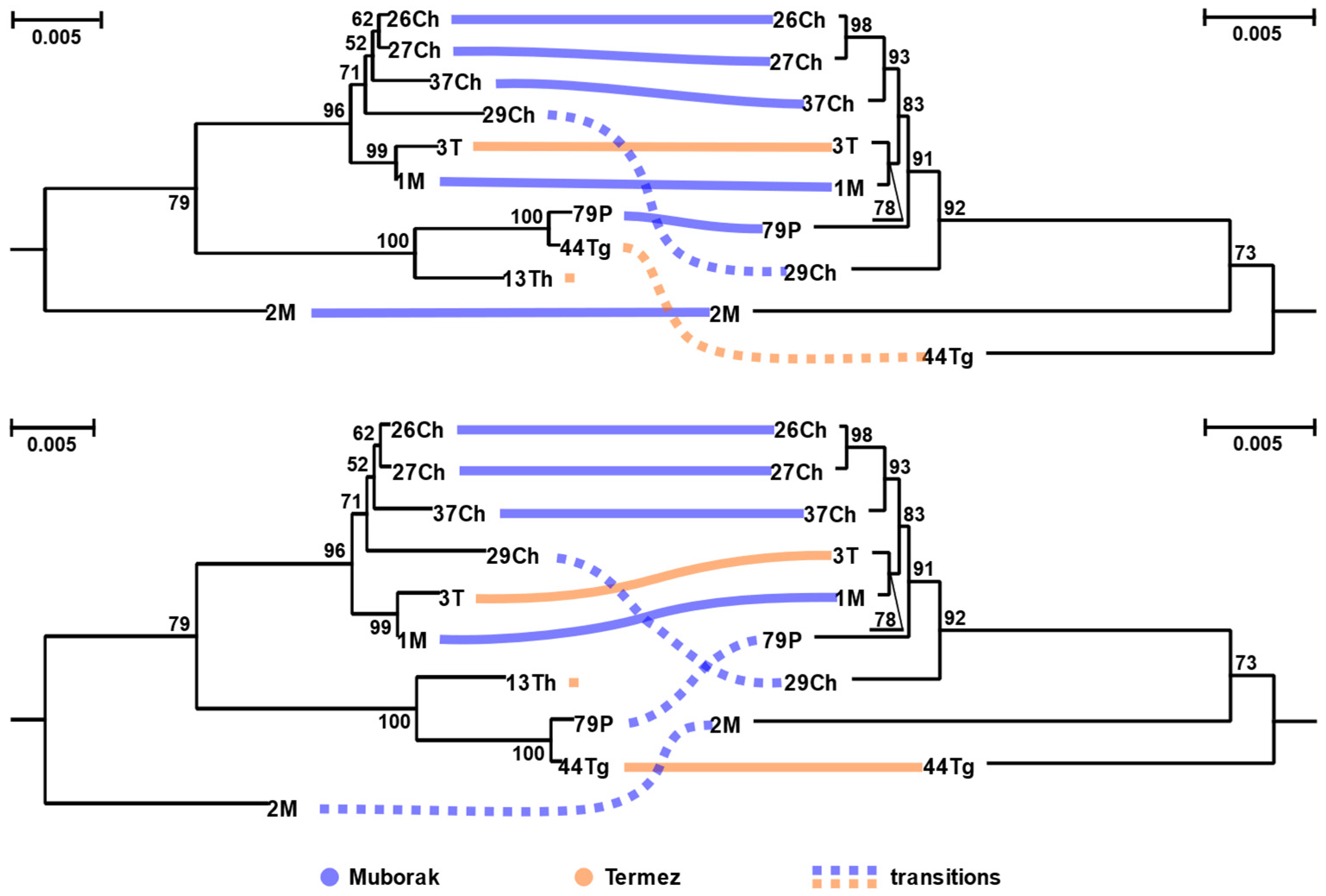
3.2. Phylogenetic Analyses of Leishmania major Strains and Their Viruses
3.3. Mode of RDRP and Capsid Genes Evolution
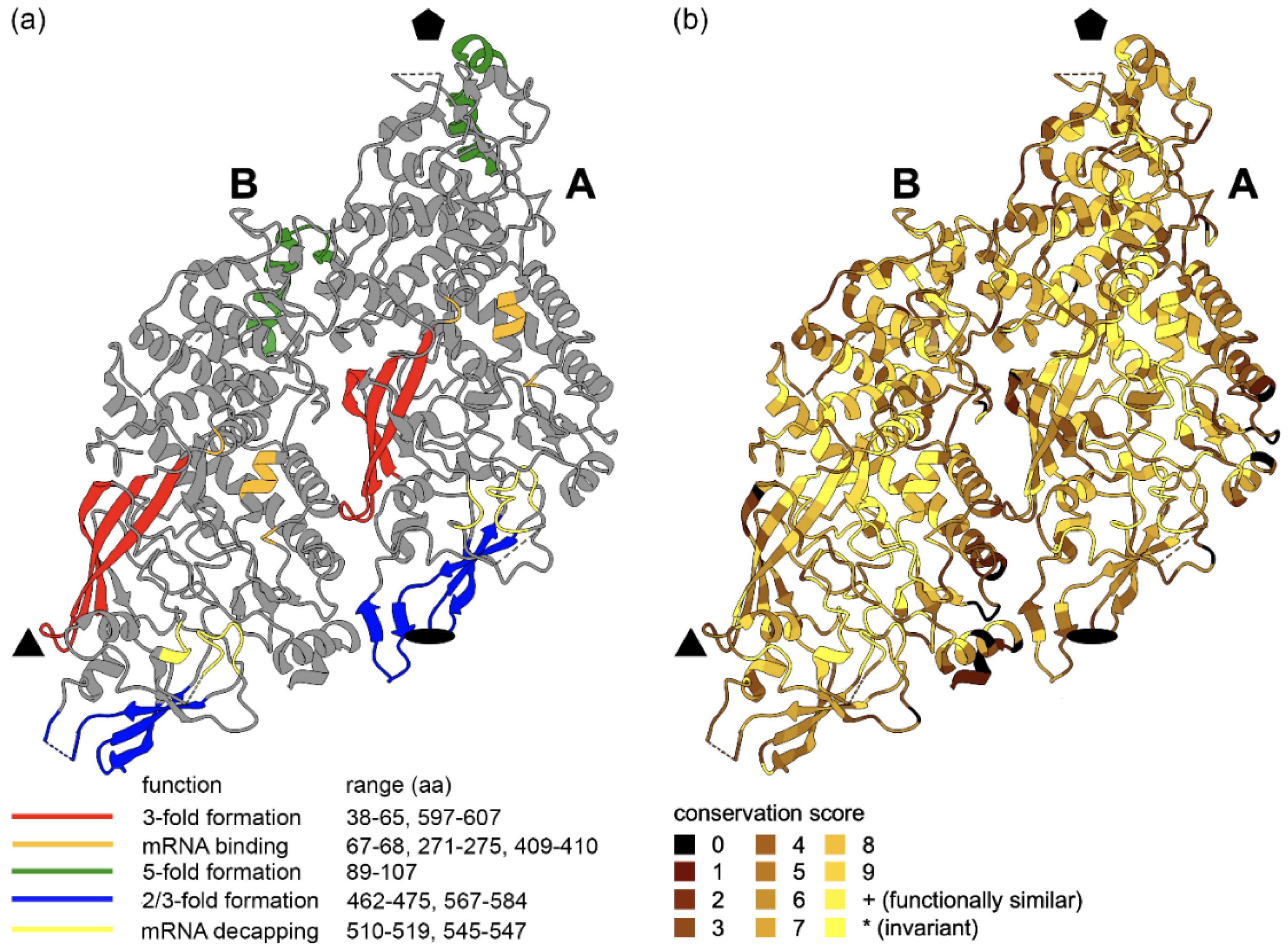
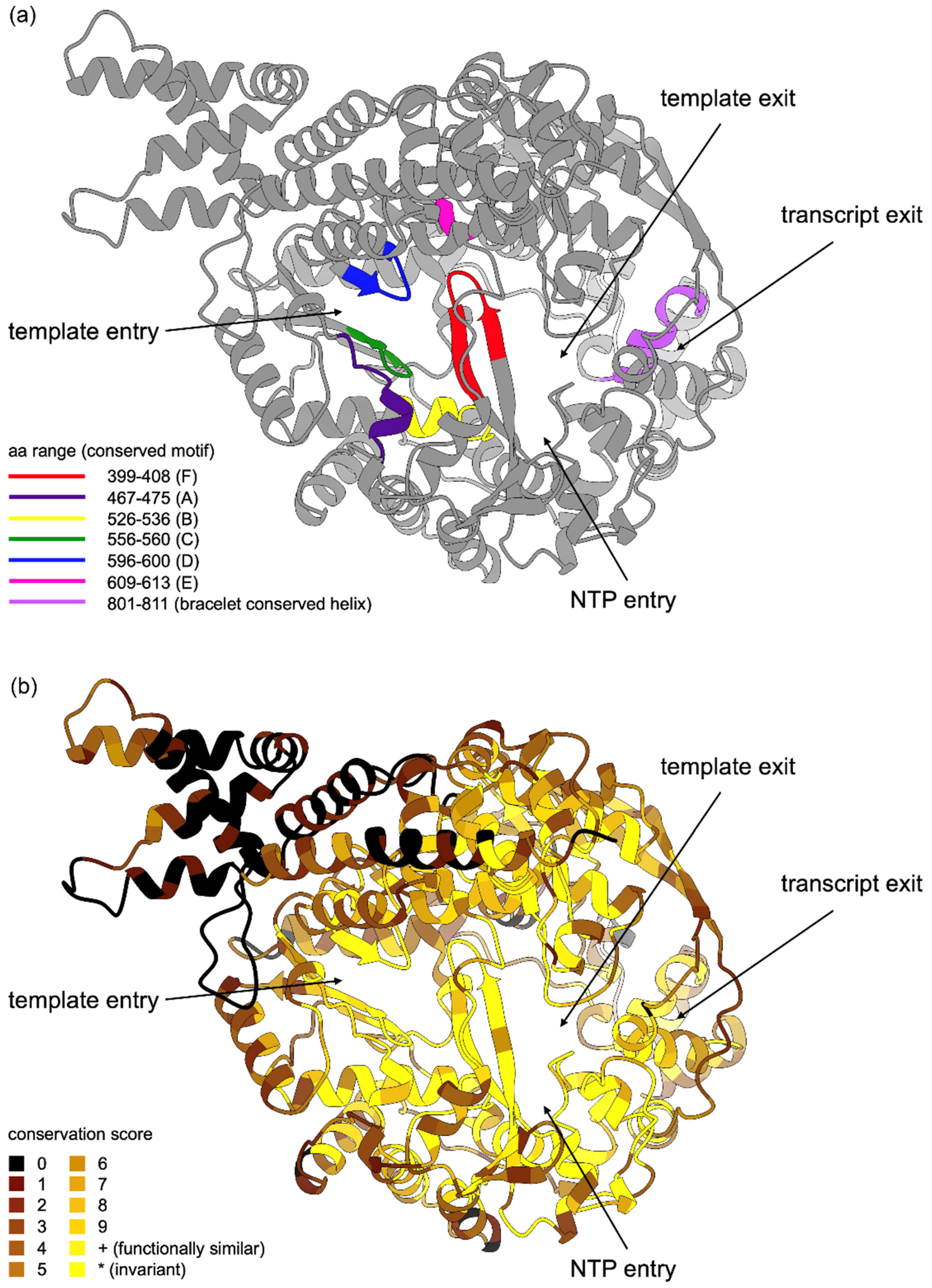
3.4. Sequence Conservation of LRV Proteins across the Structure
3.4.1. Capsid
3.4.2. RDRP
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Leishmaniasis. 2020. Available online: https://www.who.int/en/news-room/fact-sheets/detail/leishmaniasis (accessed on 12 October 2021).
- Akhoundi, M.; Downing, T.; Votýpka, J.; Kuhls, K.; Lukeš, J.; Cannet, A.; Ravel, C.; Marty, P.; Delaunay, P.; Kasbari, M.; et al. Leishmania infections: Molecular targets and diagnosis. Mol. Asp. Med. 2017, 57, 1–29. [Google Scholar] [CrossRef]
- Widmer, G.; Dooley, S. Phylogenetic analysis of Leishmania RNA virus and Leishmania suggests ancient virus-parasite association. Nucleic Acids Res. 1995, 23, 2300–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grybchuk, D.; Macedo, D.H.; Kleschenko, Y.; Kraeva, N.; Lukashev, A.N.; Bates, P.A.; Kulich, P.; Leštinová, T.; Volf, P.; Kostygov, A.Y.; et al. The first non-LRV RNA virus in Leishmania. Viruses 2020, 12, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grybchuk, D.; Akopyants, N.S.; Kostygov, A.Y.; Konovalovas, A.; Lye, L.F.; Dobson, D.E.; Zangger, H.; Fasel, N.; Butenko, A.; Frolov, A.O.; et al. Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite Leishmania. Proc. Natl. Acad. Sci. USA 2018, 115, E506–E515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghabrial, S.A.; Castón, J.R.; Jiang, D.; Nibert, M.L.; Suzuki, N. 50-plus years of fungal viruses. Virology 2015, 479–480, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Poulos, B.T.; Tang, K.F.; Pantoja, C.R.; Bonami, J.R.; Lightner, D.V. Purification and characterization of infectious myonecrosis virus of penaeid shrimp. J. Gen. Virol. 2006, 87, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Attoui, H.; Mohd Jaafar, F.; Wang, H.Q.; Cao, Y.X.; Fan, S.P.; Sun, Y.X.; Liu, L.D.; Mertens, P.P.; Meng, W.S.; et al. Isolation and full-length sequence analysis of Armigeres subalbatus totivirus, the first totivirus isolate from mosquitoes representing a proposed novel genus (Artivirus) of the family Totiviridae. J. Gen. Virol. 2010, 91, 2836–2845. [Google Scholar] [CrossRef] [PubMed]
- Løvoll, M.; Wiik-Nielsen, J.; Grove, S.; Wiik-Nielsen, C.R.; Kristoffersen, A.B.; Faller, R.; Poppe, T.; Jung, J.; Pedamallu, C.S.; Nederbragt, A.J.; et al. A novel totivirus and piscine reovirus (PRV) in Atlantic salmon (Salmo salar) with cardiomyopathy syndrome (CMS). Virol. J. 2010, 7, 309. [Google Scholar] [CrossRef] [Green Version]
- Tengs, T.; Böckerman, I. A strain of piscine myocarditis virus infecting Atlantic argentine, Argentina silus (Ascanius). J. Fish Dis. 2012, 35, 545–547. [Google Scholar] [CrossRef]
- Xin, C.; Wu, B.; Li, J.; Gong, P.; Yang, J.; Li, H.; Cai, X.; Zhang, X. Complete genome sequence and evolution analysis of Eimeria stiedai RNA virus 1, a novel member of the family Totiviridae. Arch. Virol. 2016, 161, 3571–3576. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.E.; Takagi, Y.; Parent, K.N.; Cardone, G.; Nibert, M.L.; Baker, T.S. Three-dimensional structure of a protozoal double-stranded RNA virus that infects the enteric pathogen Giardia lamblia. J. Virol. 2015, 89, 1182–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, K.N.; Takagi, Y.; Cardone, G.; Olson, N.H.; Ericsson, M.; Yang, M.; Lee, Y.; Asara, J.M.; Fichorova, R.N.; Baker, T.S.; et al. Structure of a protozoan virus from the human genitourinary parasite Trichomonas vaginalis. mBio 2013, 4, e00056-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grybchuk, D.; Kostygov, A.Y.; Macedo, D.H.; d’Avila-Levy, C.M.; Yurchenko, V. RNA viruses in trypanosomatid parasites: A historical overview. Mem. Inst. Oswaldo Cruz 2018, 113, e170487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffter, S.; Widmer, G.; Patterson, J.L. Complete sequence of Leishmania RNA virus 1–4 and identification of conserved sequences. Virology 1994, 199, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Stuart, K.D.; Weeks, R.; Guilbride, L.; Myler, P.J. Molecular organization of Leishmania RNA virus 1. Proc. Natl. Acad. Sci. USA 1992, 89, 8596–8600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantanhêde, L.M.; Mata-Somarribas, C.; Chourabi, K.; Pereira da Silva, G.; Dias das Chagas, B.; de Oliveira, R.P.L.; Cortes Boite, M.; Cupolillo, E. The maze pathway of coevolution: A critical review over the Leishmania and its endosymbiotic history. Genes 2021, 12, 657. [Google Scholar] [CrossRef]
- Grybchuk, D.; Kostygov, A.Y.; Macedo, D.H.; Votypka, J.; Lukes, J.; Yurchenko, V. RNA viruses in Blechomonas (Trypanosomatidae) and evolution of Leishmaniavirus. mBio 2018, 9, e01932-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, M.A.; Ronet, C.; Zangger, H.; Beverley, S.M.; Fasel, N. Leishmania RNA virus: When the host pays the toll. Front. Cell Infect. Microbiol. 2012, 2, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettmann, E.A.; Shaik, J.S.; Zangger, H.; Lye, L.F.; Kuhlmann, F.M.; Akopyants, N.S.; Oschwald, D.M.; Owens, K.L.; Hickerson, S.M.; Ronet, C.; et al. Tilting the balance between RNA interference and replication eradicates Leishmania RNA virus 1 and mitigates the inflammatory response. Proc. Natl. Acad. Sci. USA 2016, 113, 11998–12005. [Google Scholar] [CrossRef] [Green Version]
- Ives, A.; Ronet, C.; Prevel, F.; Ruzzante, G.; Fuertes-Marraco, S.; Schutz, F.; Zangger, H.; Revaz-Breton, M.; Lye, L.F.; Hickerson, S.M.; et al. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 2011, 331, 775–778. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Castiglioni, P.; Hartley, M.A.; Eren, R.O.; Prevel, F.; Desponds, C.; Utzschneider, D.T.; Zehn, D.; Cusi, M.G.; Kuhlmann, F.M.; et al. Type I interferons induced by endogenous or exogenous viral infections promote metastasis and relapse of leishmaniasis. Proc. Natl. Acad. Sci. USA 2017, 114, 4987–4992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, M.A.; Bourreau, E.; Rossi, M.; Castiglioni, P.; Eren, R.O.; Prevel, F.; Couppie, P.; Hickerson, S.M.; Launois, P.; Beverley, S.M.; et al. Leishmaniavirus-dependent metastatic leishmaniasis is prevented by blocking IL-17A. PLoS Pathog. 2016, 12, e1005852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zangger, H.; Hailu, A.; Desponds, C.; Lye, L.F.; Akopyants, N.S.; Dobson, D.E.; Ronet, C.; Ghalib, H.; Beverley, S.M.; Fasel, N. Leishmania aethiopica field isolates bearing an endosymbiontic dsRNA virus induce pro-inflammatory cytokine response. PLoS Negl. Trop. Dis. 2014, 8, e2836. [Google Scholar] [CrossRef] [Green Version]
- Eren, R.O.; Reverte, M.; Rossi, M.; Hartley, M.A.; Castiglioni, P.; Prevel, F.; Martin, R.; Desponds, C.; Lye, L.F.; Drexler, S.K.; et al. Mammalian innate immune response to a Leishmania-resident RNA virus increases macrophage survival to promote parasite persistence. Cell Host Microbe 2016, 20, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrow, P.; Dujardin, J.C.; Fasel, N.; Greenwood, A.D.; Osterrieder, K.; Lomonossoff, G.; Fiori, P.L.; Atterbury, R.; Rossi, M.; Lalle, M. Viruses of protozoan parasites and viral therapy: Is the time now right? Virol. J. 2020, 17, 142. [Google Scholar] [CrossRef]
- Macedo, D.H.; Menezes-Neto, A.; Rugani, J.M.; Rocha, A.C.; Silva, S.O.; Melo, M.N.; Lye, L.F.; Beverley, S.M.; Gontijo, C.M.; Soares, R.P. Low frequency of LRV1 in Leishmania braziliensis strains isolated from typical and atypical lesions in the State of Minas Gerais, Brazil. Mol. Biochem. Parasitol. 2016, 210, 50–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajjaran, H.; Mahdi, M.; Mohebali, M.; Samimi-Rad, K.; Ataei-Pirkooh, A.; Kazemi-Rad, E.; Naddaf, S.R.; Raoofian, R. Detection and molecular identification of Leishmania RNA virus (LRV) in Iranian Leishmania species. Arch. Virol. 2016, 161, 3385–3390. [Google Scholar] [CrossRef]
- Saberi, R.; Fakhar, M.; Hajjaran, H.; Ataei-Pirkooh, A.; Mohebali, M.; Taghipour, N.; Ziaei Hezarjaribi, H.; Moghadam, Y.; Bagheri, A. Presence and diversity of Leishmania RNA virus in an old zoonotic cutaneous leishmaniasis focus, northeastern Iran: Haplotype and phylogenetic based approach. Int. J. Infect. Dis. 2020, 101, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Atayde, V.D.; da Silva Lira Filho, A.; Chaparro, V.; Zimmermann, A.; Martel, C.; Jaramillo, M.; Olivier, M. Exploitation of the Leishmania exosomal pathway by Leishmania RNA virus 1. Nat. Microbiol. 2019, 4, 714–723. [Google Scholar] [CrossRef]
- Ishemgulova, A.; Kraeva, N.; Hlaváčová, J.; Zimmer, S.L.; Butenko, A.; Podešvová, L.; Leštinová, T.; Lukeš, J.; Kostygov, A.; Votýpka, J.; et al. A putative ATP/GTP binding protein affects Leishmania mexicana growth in insect vectors and vertebrate hosts. PLoS Negl. Trop. Dis. 2017, 11, e0005782. [Google Scholar] [CrossRef] [Green Version]
- Maslov, D.A.; Lukeš, J.; Jirků, M.; Simpson, L. Phylogeny of trypanosomes as inferred from the small and large subunit rRNAs: Implications for the evolution of parasitism in the trypanosomatid protozoa. Mol. Biochem. Parasitol. 1996, 75, 197–205. [Google Scholar] [CrossRef]
- Losev, A.; Grybchuk-Ieremenko, A.; Kostygov, A.Y.; Lukes, J.; Yurchenko, V. Host specificity, pathogenicity, and mixed infections of trypanoplasms from freshwater fishes. Parasitol. Res. 2015, 114, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Kleschenko, Y.; Grybchuk, D.; Matveeva, N.S.; Macedo, D.H.; Ponirovsky, E.N.; Lukashev, A.N.; Yurchenko, V. Molecular characterization of Leishmania RNA virus 2 in Leishmania major from Uzbekistan. Genes 2019, 10, 830. [Google Scholar] [CrossRef] [Green Version]
- Sádlová, J.; Podešvová, L.; Bečvář, T.; Bianchi, C.; Gerasimov, E.S.; Saura, A.; Glanzová, K.; Leštinová, T.; Matveeva, N.S.; Chmelová, L.; et al. Catalase impairs Leishmania mexicana development and virulence. Virulence 2021, 12, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 8 January 2019. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 12 October 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Aslett, M.; Aurrecoechea, C.; Berriman, M.; Brestelli, J.; Brunk, B.P.; Carrington, M.; Depledge, D.P.; Fischer, S.; Gajria, B.; Gao, X.; et al. TriTrypDB: A functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010, 38, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2020; p. 496. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodcroft, B.J.; Boyd, J.A.; Tyson, G.W. OrfM: A fast open reading frame predictor for metagenomic data. Bioinformatics 2016, 32, 2702–2703. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, J. A simple method for estimating the strength of natural selection on overlapping genes. Genome Biol. Evol. 2014, 7, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Massingham, T.; Goldman, N. Detecting amino acid sites under positive selection and purifying selection. Genetics 2005, 169, 1753–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Procházková, M.; Füzik, T.; Grybchuk, D.; Falginella, F.L.; Podešvová, L.; Yurchenko, V.; Vácha, R.; Plevka, P. Capsid structure of Leishmania RNA Virus 1. J. Virol. 2021, 95, e01957-20. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Bruenn, J.A. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res. 2003, 31, 1821–1829. [Google Scholar] [CrossRef]
- Procter, J.B.; Carstairs, G.M.; Soares, B.; Mourao, K.; Ofoegbu, T.C.; Barton, D.; Lui, L.; Menard, A.; Sherstnev, N.; Roldan-Martinez, D.; et al. Alignment of biological sequences with Jalview. Methods Mol. Biol. 2021, 2231, 203–224. [Google Scholar] [PubMed]
- Naitow, H.; Tang, J.; Canady, M.; Wickner, R.B.; Johnson, J.E. L-A virus at 3.4 A resolution reveals particle architecture and mRNA decapping mechanism. Nat. Struct. Biol. 2002, 9, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, S.; Prasad, B.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from structure, function and evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Zhang, Y.; Zhou, K.; Sun, J.; Zhou, Z.H. Conservative transcription in three steps visualized in a double-stranded RNA virus. Nat. Struct. Mol. Biol. 2019, 26, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Celma, C.C.; Zhang, X.; Chang, T.; Shen, W.; Atanasov, I.; Roy, P.; Zhou, Z.H. In situ structures of rotavirus polymerase in action and mechanism of mRNA transcription and release. Nat. Commun. 2019, 10, 2216. [Google Scholar] [CrossRef]
- Lawton, J.A.; Estes, M.K.; Prasad, B.V. Mechanism of genome transcription in segmented dsRNA viruses. Adv. Virus Res. 2000, 55, 185–229. [Google Scholar] [PubMed]
- Butenko, A.; Kostygov, A.Y.; Sádlová, J.; Kleschenko, Y.; Bečvář, T.; Podešvová, L.; Macedo, D.H.; Žihala, D.; Lukeš, J.; Bates, P.A.; et al. Comparative genomics of Leishmania (Mundinia). BMC Genom. 2019, 20, 726. [Google Scholar] [CrossRef] [Green Version]
- Harkins, K.M.; Schwartz, R.S.; Cartwright, R.A.; Stone, A.C. Phylogenomic reconstruction supports supercontinent origins for Leishmania. Infect. Genet. Evol. 2016, 38, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Chajbullinova, A.; Votýpka, J.; Sádlová, J.; Kvapilová, K.; Seblová, V.; Kreisinger, J.; Jirků, M.; Sanjoba, C.; Gantuya, S.; Matsumoto, Y.; et al. The development of Leishmania turanica in sand flies and competition with L. major. Parasit. Vectors 2012, 5, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strelkova, M.V.; Eliseev, L.N.; Ponirovsky, E.N.; Dergacheva, T.I.; Annacharyeva, D.K.; Erokhin, P.I.; Evans, D.A. Mixed leishmanial infections in Rhombomys opimus: A key to the persistence of Leishmania major from one transmission season to the next. Ann. Trop. Med. Parasitol. 2001, 95, 811–819. [Google Scholar] [PubMed]
- Nalçacı, M.; Karakuş, M.; Yilmaz, B.; Demir, S.; Özbilgin, A.; Özbel, Y.; Töz, S. Detection of Leishmania RNA virus 2 in Leishmania species from Turkey. Trans. R. Soc. Trop. Med. Hyg. 2019, 113, 410–417. [Google Scholar] [CrossRef]
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votýpka, J.; Marty, P.; Delaunay, P.; Sereno, D. A historical overview of the classification, evolution, and dispersion of Leishmania parasites and sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef] [PubMed]
- Tibayrenc, M.; Ayala, F.J. Models in parasite and pathogen evolution: Genomic analysis reveals predominant clonality and progressive evolution at all evolutionary scales in parasitic protozoa, yeasts and bacteria. Adv. Parasitol. 2021, 111, 75–117. [Google Scholar] [PubMed]
- Akopyants, N.S.; Kimblin, N.; Secundino, N.; Patrick, R.; Peters, N.; Lawyer, P.; Dobson, D.E.; Beverley, S.M.; Sacks, D.L. Demonstration of genetic exchange during cyclical development of Leishmania in the sand fly vector. Science 2009, 324, 265–268. [Google Scholar] [CrossRef] [Green Version]
- Volf, P.; Sádlová, J. Sex in Leishmania. Science 2009, 324, 1644. [Google Scholar] [CrossRef]
- Gutiérrez-Corbo, C.; Dominguez-Asenjo, B.; Martinez-Valladares, M.; Pérez-Pertejo, Y.; García-Estrada, C.; Balaña-Fouce, R.; Reguera, R.M. Reproduction in trypanosomatids: Past and present. Biology 2021, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Tirera, S.; Ginouves, M.; Donato, D.; Caballero, I.S.; Bouchier, C.; Lavergne, A.; Bourreau, E.; Mosnier, E.; Vantilcke, V.; Couppie, P.; et al. Unraveling the genetic diversity and phylogeny of Leishmania RNA virus 1 strains of infected Leishmania isolates circulating in French Guiana. PLoS Negl. Trop. Dis. 2017, 11, e0005764. [Google Scholar] [CrossRef] [PubMed]
- Matveyev, A.V.; Alves, J.M.; Serrano, M.G.; Lee, V.; Lara, A.M.; Barton, W.A.; Costa-Martins, A.G.; Beverley, S.M.; Camargo, E.P.; Teixeira, M.M.; et al. The evolutionary loss of RNAi key determinants in kinetoplastids as a multiple sporadic phenomenon. J. Mol. Evol. 2017, 84, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Lye, L.F.; Owens, K.; Shi, H.; Murta, S.M.; Vieira, A.C.; Turco, S.J.; Tschudi, C.; Ullu, E.; Beverley, S.M. Retention and loss of RNA interference pathways in trypanosomatid protozoans. PLoS Pathog. 2010, 6, e1001161. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Code | WHO Code | Source | Origin |
|---|---|---|---|---|
| Negative | ||||
| L. turanica | 87568 | MRHO/UZ/1987/KD87568 | great gerbil 1 | Uzbekistan, Qashqadaryo reg |
| 9554 | MRHO/TM/1995/9554 | Turkmenistan, Ak bugdaý, Ahal reg. | ||
| 9562 | MRHO/TM/1995/9562 | |||
| 9558 | MRHO/TM/1995/9558 | |||
| KP137 | MHOM/TM/1986/KP137 | Turkmenistan, Serdar, Balkan reg. | ||
| 91014 | MHOM/TM/1991/91014 | Turkmenistan, Tjazeel, Ahal reg. | ||
| 73 P | MHOM/UZ/2002/IsvM73g | Uzbekistan, Muborak, Qashqadaryo reg. | ||
| 9105 | MRHO/TM/1991/9105 | Turkmenistan, Tjazeel, Ahal reg. | ||
| L. major | 13Th | MHOM/UZ/2003/IsvT13h | human | Uzbekistan, Termez, Surxondaryo reg. |
| 24Th | MRHO/UZ/2003/IsvT24h | |||
| 9537 | MRHO/TM/1995/9537 | great gerbil | Turkmenistan, Serahs, Ahal reg. | |
| BUR | MRHO/UZ/1987/BUR | human | Uzbekistan, Qorovulbozor, Bukhara reg. | |
| Positive | ||||
| L. major | 1M 2 | MHOM/UZ/1998/IsvM01h | human | Uzbekistan, Muborak, Qashqadaryo reg. |
| 2M | MHOM/UZ/1998/IsvM29h | |||
| 26Ch | MHOM/UZ/2002/IsvM26h | |||
| 27Ch 2 | MHOM/UZ/1998/IsvM27h | |||
| 29Ch | MHOM/UZ/2002/IsvM29h | |||
| 37Ch | MHOM/UZ/2002/IsvM37h | |||
| 79P | MRHO/UZ/2002/IsvM79g | great gerbil | ||
| 44Tg | MRHO/UZ/2003/IsvT44g | Uzbekistan, Termez, Surxondaryo reg. | ||
| 3T | MHOM/UZ/2000/IsvT03h | human | ||
| RDRP | Capsid | |
|---|---|---|
| LRV1 | 0.0617 | 0.0219 |
| LRV2 from L. major | 0.1283 | 0.0292 |
| LRV2 from L. aethiopica | 0.0455 | 0.0116 |
| overall | 0.0554 | 0.0192 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostygov, A.Y.; Grybchuk, D.; Kleschenko, Y.; Chistyakov, D.S.; Lukashev, A.N.; Gerasimov, E.S.; Yurchenko, V. Analyses of Leishmania-LRV Co-Phylogenetic Patterns and Evolutionary Variability of Viral Proteins. Viruses 2021, 13, 2305. https://doi.org/10.3390/v13112305
Kostygov AY, Grybchuk D, Kleschenko Y, Chistyakov DS, Lukashev AN, Gerasimov ES, Yurchenko V. Analyses of Leishmania-LRV Co-Phylogenetic Patterns and Evolutionary Variability of Viral Proteins. Viruses. 2021; 13(11):2305. https://doi.org/10.3390/v13112305
Chicago/Turabian StyleKostygov, Alexei Y., Danyil Grybchuk, Yulia Kleschenko, Daniil S. Chistyakov, Alexander N. Lukashev, Evgeny S. Gerasimov, and Vyacheslav Yurchenko. 2021. "Analyses of Leishmania-LRV Co-Phylogenetic Patterns and Evolutionary Variability of Viral Proteins" Viruses 13, no. 11: 2305. https://doi.org/10.3390/v13112305
APA StyleKostygov, A. Y., Grybchuk, D., Kleschenko, Y., Chistyakov, D. S., Lukashev, A. N., Gerasimov, E. S., & Yurchenko, V. (2021). Analyses of Leishmania-LRV Co-Phylogenetic Patterns and Evolutionary Variability of Viral Proteins. Viruses, 13(11), 2305. https://doi.org/10.3390/v13112305