
Eurasian Avian-like M1 Plays More Important Role than M2 in Pathogenicity of 2009 Pandemic H1N1 Influenza Virus in Mice


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
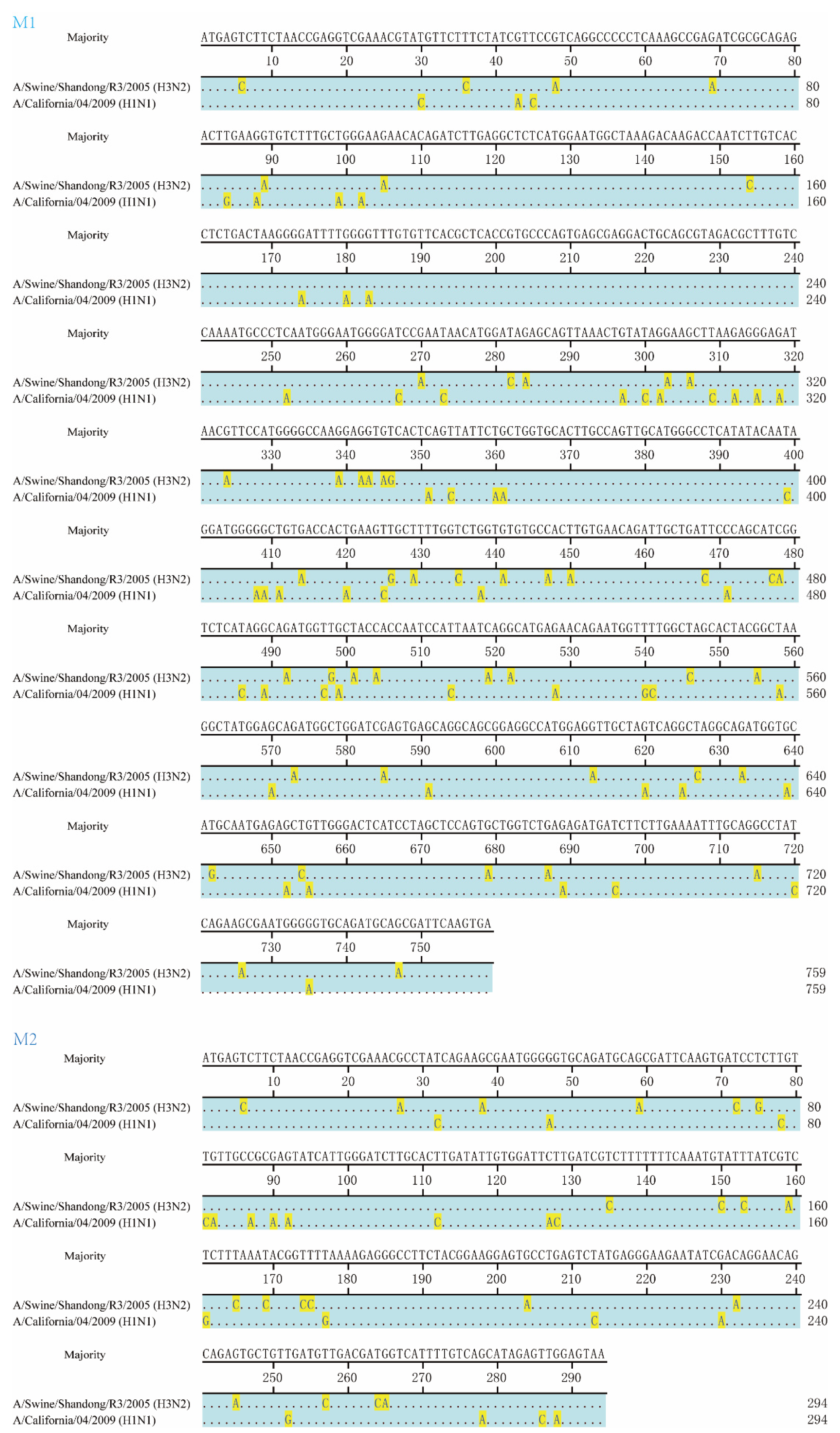
2.2. Construction of Plasmids
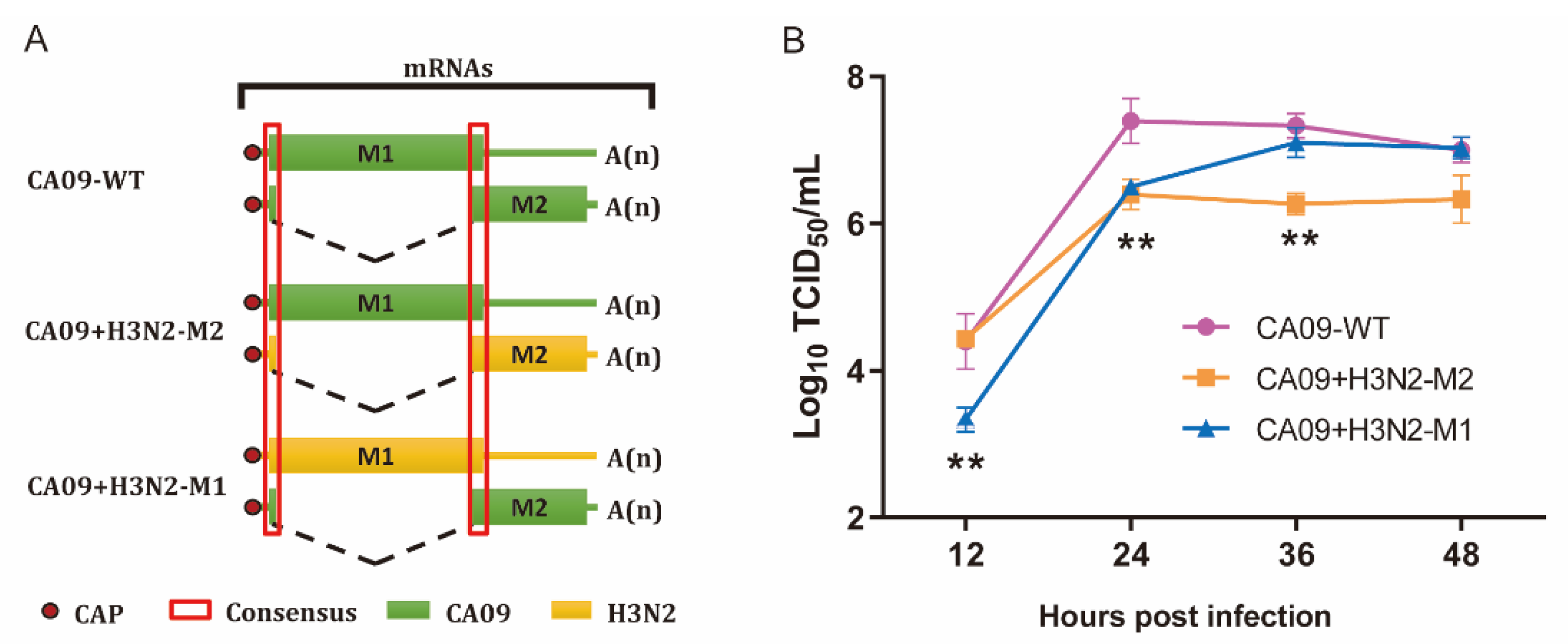
2.3. Rescue of Recombinant Influenza a Viruses
2.4. Virus Growth Kinetic Assay
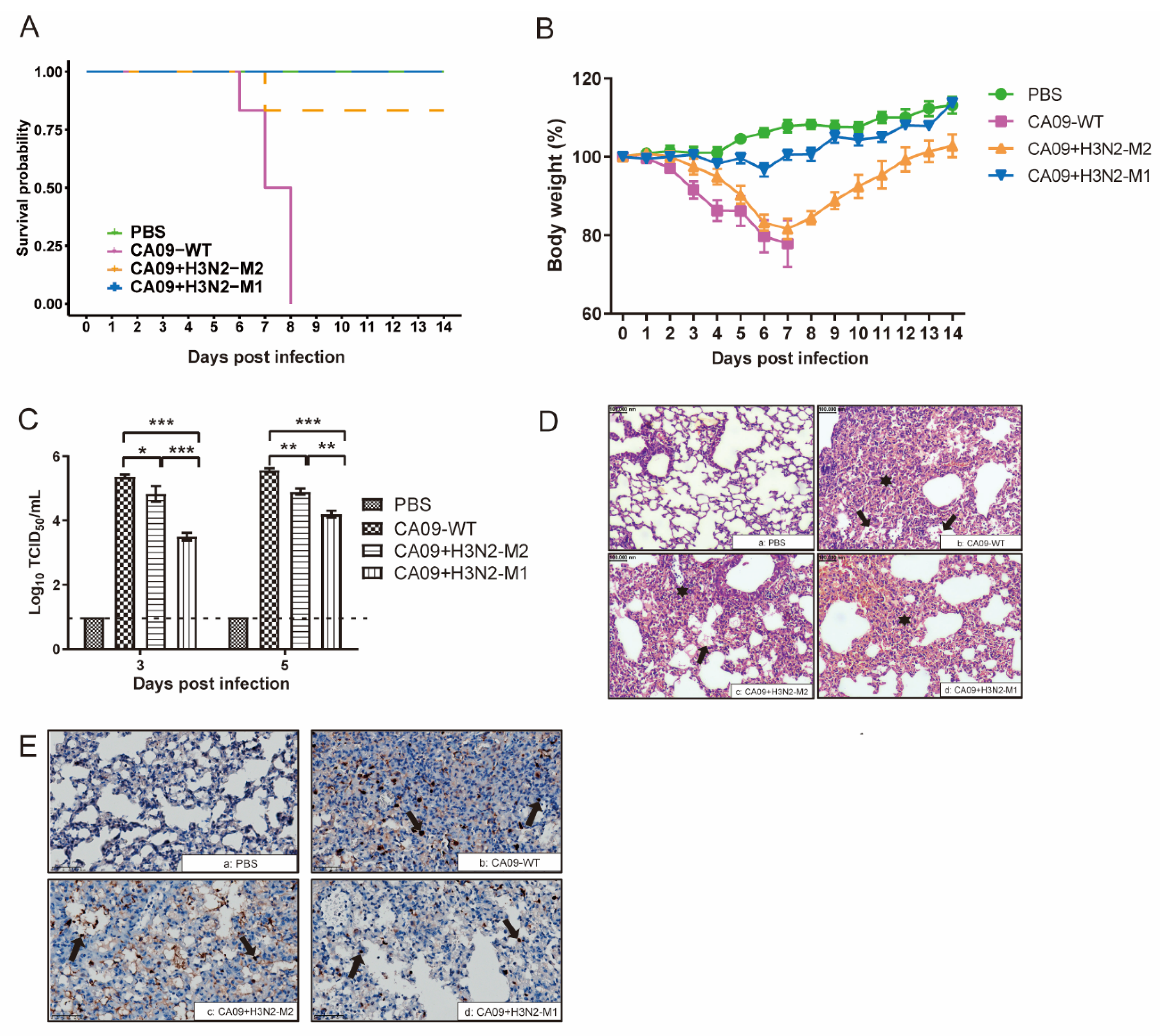
2.5. Animal Study
2.6. Determination of Cytokine Levels in Mouse Lungs
2.7. Ethics Statement and Statistical Analysis
3. Results
3.1. M1 and M2 of H3N2 Affected Replication of Chimeric Viruses In Vitro
3.2. H3N2 M1 Attenuated CA09 + H3N2-M1 in Mice
3.3. CA09 + H3N2-M1 Induced Lower Inflammatory Responses, whereas CA09 + H3N2-M2 Induced Comparable Inflammatory Responses with Wild Virus
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Influenza A(H1N1)—Update 61; WHO: Geneva, Switzerland, 2009. [Google Scholar]
- Kasowski, E.J.; Garten, R.J.; Bridges, C.B. Influenza pandemic epidemiologic and virologic diversity: Reminding ourselves of the possibilities. Clin. Infect. Dis. 2011, 52 (Suppl. S1), S44–S49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Wei, Z.; Greene, C.M.; Yang, P.; Su, J.; Song, Y.; Iuliano, A.D.; Wang, Q. Mortality burden from seasonal influenza and 2009 H1N1 pandemic influenza in Beijing, China, 2007–2013. Influenza Other Respir. Viruses 2018, 12, 88–97. [Google Scholar] [CrossRef]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.; Dennis, A.; Flutter, C.; Khan, Z. Pandemic (H1N1) 2009 influenza. Br. J. Anaesth. 2010, 104, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Sun, S.; Du, L.; Ma, J.; Fan, L.; Pu, J.; Sun, Y.; Zhao, J.; Sun, H.; Liu, J. Natural and experimental infection of dogs with pandemic H1N1/2009 influenza virus. J. Gen. Virol. 2012, 93, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Pantin-Jackwood, M.; Wasilenko, J.L.; Spackman, E.; Suarez, D.L.; Swayne, D.E. Susceptibility of turkeys to pandemic-H1N1 virus by reproductive tract insemination. Virol. J. 2010, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Fiorentini, L.; Taddei, R.; Moreno, A.; Gelmetti, D.; Barbieri, I.; De Marco, M.A.; Tosi, G.; Cordioli, P.; Massi, P. Influenza A pandemic (H1N1) 2009 virus outbreak in a cat colony in Italy. Zoonoses Public Health 2011, 58, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Howden, K.J.; Brockhoff, E.J.; Caya, F.D.; McLeod, L.J.; Lavoie, M.; Ing, J.D.; Bystrom, J.M.; Alexandersen, S.; Pasick, J.M.; Berhane, Y.; et al. An investigation into human pandemic influenza virus (H1N1) 2009 on an Alberta swine farm. Can. Vet. J. 2009, 50, 1153–1161. [Google Scholar] [PubMed]
- Schrenzel, M.D.; Tucker, T.A.; Stalis, I.H.; Kagan, R.A.; Burns, R.P.; Denison, A.M.; Drew, C.P.; Paddock, C.D.; Rideout, B.A. Pandemic (H1N1) 2009 virus in 3 wildlife species, San Diego, California, USA. Emerg. Infect. Dis. 2011, 17, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Welsh, M.D.; Baird, P.M.; Guelbenzu-Gonzalo, M.P.; Hanna, A.; Reid, S.M.; Essen, S.; Russell, C.; Thomas, S.; Barrass, L.; McNeilly, F.; et al. Initial incursion of pandemic (H1N1) 2009 influenza A virus into European pigs. Vet. Rec. 2010, 166, 642–645. [Google Scholar] [CrossRef]
- Sreta, D.; Tantawet, S.; Na Ayudhya, S.N.; Thontiravong, A.; Wongphatcharachai, M.; Lapkuntod, J.; Bunpapong, N.; Tuanudom, R.; Suradhat, S.; Vimolket, L.; et al. Pandemic (H1N1) 2009 virus on commercial swine farm, Thailand. Emerg. Infect. Dis. 2010, 16, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Ducatez, M.F.; Hause, B.; Stigger-Rosser, E.; Darnell, D.; Corzo, C.; Juleen, K.; Simonson, R.; Brockwell-Staats, C.; Rubrum, A.; Wang, D.; et al. Multiple reassortment between pandemic (H1N1) 2009 and endemic influenza viruses in pigs, United States. Emerg. Infect. Dis. 2011, 17, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Epperson, S.; Jhung, M.; Richards, S.; Quinlisk, P.; Ball, L.; Moll, M.; Boulton, R.; Haddy, L.; Biggerstaff, M.; Brammer, L.; et al. Human infections with influenza A(H3N2) variant virus in the United States, 2011–2012. Clin. Infect. Dis. 2013, 57 (Suppl. S1), S4–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhung, M.A.; Epperson, S.; Biggerstaff, M.; Allen, D.; Balish, A.; Barnes, N.; Beaudoin, A.; Berman, L.; Bidol, S.; Blanton, L.; et al. Outbreak of variant influenza A(H3N2) virus in the United States. Clin. Infect. Dis. 2013, 57, 1703–1712. [Google Scholar] [CrossRef] [Green Version]
- Dawood, F.S.; Jain, S.; Finelli, L.; Shaw, M.W.; Lindstrom, S.; Garten, R.J.; Gubareva, L.V.; Xu, X.; Bridges, C.B.; Uyeki, T.M. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med. 2009, 360, 2605–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, V.; Bridges, C.B.; Uyeki, T.M.; Shu, B.; Balish, A.; Xu, X.; Lindstrom, S.; Gubareva, L.V.; Deyde, V.; Garten, R.J.; et al. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005–2009. N. Engl. J. Med. 2009, 360, 2616–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Wang, G.; Mo, Y.; Yu, Q.; Xiao, X.; Yang, W.; Zhao, W.; Guo, X.; Chen, Q.; He, J.; et al. Novel triple-reassortant influenza viruses in pigs, Guangxi, China. Emerg. Microbes Infect. 2018, 7, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajao, D.S.; Walia, R.R.; Campbell, B.; Gauger, P.C.; Janas-Martindale, A.; Killian, M.L.; Vincent, A.L. Reassortment between Swine H3N2 and 2009 Pandemic H1N1 in the United States Resulted in Influenza A Viruses with Diverse Genetic Constellations with Variable Virulence in Pigs. J. Virol. 2017, 91, e01763-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakdawala, S.S.; Lamirande, E.W.; Suguitan, A.L., Jr.; Wang, W.; Santos, C.P.; Vogel, L.; Matsuoka, Y.; Lindsley, W.G.; Jin, H.; Subbarao, K. Eurasian-origin gene segments contribute to the transmissibility, aerosol release, and morphology of the 2009 pandemic H1N1 influenza virus. PLoS Pathog. 2011, 7, e1002443. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.Y.; Albrecht, R.A.; Pica, N.; Lowen, A.C.; Richt, J.A.; Garcia-Sastre, A.; Palese, P.; Hai, R. The M segment of the 2009 new pandemic H1N1 influenza virus is critical for its high transmission efficiency in the guinea pig model. J. Virol. 2011, 85, 11235–11241. [Google Scholar] [CrossRef] [Green Version]
- Calderon, B.M.; Danzy, S.; Delima, G.K.; Jacobs, N.T.; Ganti, K.; Hockman, M.R.; Conn, G.L.; Lowen, A.C.; Steel, J. Dysregulation of M segment gene expression contributes to influenza A virus host restriction. PLoS Pathog. 2019, 15, e1007892. [Google Scholar] [CrossRef] [Green Version]
- Elleman, C.J.; Barclay, W.S. The M1 matrix protein controls the filamentous phenotype of influenza A virus. Virology 2004, 321, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; Kyriakis, C.S.; Marshall, N.; Suppiah, S.; Seladi-Schulman, J.; Danzy, S.; Lowen, A.C.; Steel, J. Residue 41 of the Eurasian avian-like swine influenza a virus matrix protein modulates virion filament length and efficiency of contact transmission. J. Virol. 2014, 88, 7569–7577. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; Danzy, S.; Kyriakis, C.S.; Deymier, M.J.; Lowen, A.C.; Steel, J. The M segment of the 2009 pandemic influenza virus confers increased neuraminidase activity, filamentous morphology, and efficient contact transmissibility to A/Puerto Rico/8/1934-based reassortant viruses. J. Virol. 2014, 88, 3802–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chizhmakov, I.V.; Geraghty, F.M.; Ogden, D.C.; Hayhurst, A.; Antoniou, M.; Hay, A.J. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J. Physiol. 1996, 494 Pt 2, 329–336. [Google Scholar] [CrossRef]
- Schnell, J.R.; Chou, J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauffer, S.; Feng, Y.; Nebioglu, F.; Heilig, R.; Picotti, P.; Helenius, A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J. Virol. 2014, 88, 13029–13046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossman, J.S.; Lamb, R.A. Influenza virus assembly and budding. Virology 2011, 411, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leser, G.P.; Lamb, R.A. Influenza virus assembly and budding in raft-derived microdomains: A quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology 2005, 342, 215–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Puertas, P.; Albo, C.; Perez-Pastrana, E.; Vivo, A.; Portela, A. Influenza virus matrix protein is the major driving force in virus budding. J. Virol. 2000, 74, 11538–11547. [Google Scholar] [CrossRef] [Green Version]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef] [Green Version]
- Martyna, A.; Rossman, J. Alterations of membrane curvature during influenza virus budding. Biochem. Soc. Trans. 2014, 42, 1425–1428. [Google Scholar] [CrossRef]
- Uraki, R.; Kiso, M.; Shinya, K.; Goto, H.; Takano, R.; Iwatsuki-Horimoto, K.; Takahashi, K.; Daniels, R.S.; Hungnes, O.; Watanabe, T.; et al. Virulence determinants of pandemic A(H1N1)2009 influenza virus in a mouse model. J. Virol. 2013, 87, 2226–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiso, M.; Shinya, K.; Shimojima, M.; Takano, R.; Takahashi, K.; Katsura, H.; Kakugawa, S.; Le, M.T.; Yamashita, M.; Furuta, Y.; et al. Characterization of oseltamivir-resistant 2009 H1N1 pandemic influenza A viruses. PLoS Pathog. 2010, 6, e1001079. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Lager, K.M.; Lekcharoensuk, P.; Ulery, E.S.; Janke, B.H.; Solorzano, A.; Webby, R.J.; Garcia-Sastre, A.; Richt, J.A. Viral reassortment and transmission after co-infection of pigs with classical H1N1 and triple-reassortant H3N2 swine influenza viruses. J. Gen. Virol. 2010, 91, 2314–2321. [Google Scholar] [CrossRef]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yu, H.; Li, Y.; Ma, J.; Lang, Y.; Duff, M.; Henningson, J.; Liu, Q.; Li, Y.; Nagy, A.; et al. Impacts of different expressions of PA-X protein on 2009 pandemic H1N1 virus replication, pathogenicity and host immune responses. Virology 2017, 504, 25–35. [Google Scholar] [CrossRef]
- Richt, J.A.; Lager, K.M.; Janke, B.H.; Woods, R.D.; Webster, R.G.; Webby, R.J. Pathogenic and antigenic properties of phylogenetically distinct reassortant H3N2 swine influenza viruses cocirculating in the United States. J. Clin. Microbiol. 2003, 41, 3198–3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhu, J.; Li, Y.; Bradley, K.C.; Cao, J.; Chen, H.; Jin, M.; Zhou, H. Glycosylation on hemagglutinin affects the virulence and pathogenicity of pandemic H1N1/2009 influenza A virus in mice. PLoS ONE 2013, 8, e61397. [Google Scholar] [CrossRef]
- Ahangar-Parvin, R.; Mohammadi-Kordkhayli, M.; Azizi, S.V.; Nemati, M.; Khorramdelazad, H.; Taghipour, Z.; Hassan, Z.; Moazzeni, S.M.; Jafarzadeh, A. The Modulatory Effects of Vitamin D on the Expression of IL-12 and TGF-beta in the Spinal Cord and Serum of Mice with Experimental Autoimmune Encephalomyelitis. Iran. J. Pathol. 2018, 13, 10–22. [Google Scholar] [PubMed]
- CDC. 2009 H1N1 Pandemic Timeline; CDC: Atlanta, GA, USA, 2019.
- Walia, R.R.; Anderson, T.K.; Vincent, A.L. Regional patterns of genetic diversity in swine influenza A viruses in the United States from 2010 to 2016. Influenza Other Respir. Viruses 2019, 13, 262–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stincarelli, M.; Arvia, R.; De Marco, M.A.; Clausi, V.; Corcioli, F.; Cotti, C.; Delogu, M.; Donatelli, I.; Azzi, A.; Giannecchini, S. Reassortment ability of the 2009 pandemic H1N1 influenza virus with circulating human and avian influenza viruses: Public health risk implications. Virus Res. 2013, 175, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Liu, Q.; Bawa, B.; Qiao, C.; Qi, W.; Shen, H.; Chen, Y.; Ma, J.; Li, X.; Webby, R.J.; et al. The neuraminidase and matrix genes of the 2009 pandemic influenza H1N1 virus cooperate functionally to facilitate efficient replication and transmissibility in pigs. J. Gen. Virol. 2012, 93, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Pulit-Penaloza, J.A.; Jones, J.; Sun, X.; Jang, Y.; Thor, S.; Belser, J.A.; Zanders, N.; Creager, H.M.; Ridenour, C.; Wang, L.; et al. Antigenically Diverse Swine Origin H1N1 Variant Influenza Viruses Exhibit Differential Ferret Pathogenesis and Transmission Phenotypes. J. Virol. 2018, 92, e00095-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, P.; Huang, M.; Qiao, S.; Liu, Q.; Chen, H.; Teng, Q.; Li, X.; Zhang, Z.; Yan, D.; et al. Key amino acids of M1-41 and M2-27 determine growth and pathogenicity of chimeric H17 bat influenza virus in cells and in mice. J. Virol. 2021, 95, e01019-21. [Google Scholar] [CrossRef] [PubMed]
- Bialas, K.M.; Desmet, E.A.; Takimoto, T. Specific residues in the 2009 H1N1 swine-origin influenza matrix protein influence virion morphology and efficiency of viral spread in vitro. PLoS ONE 2012, 7, e50595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welman, M.; Arora, D.J. Genomic analysis of matrix gene and antigenic studies of its gene product (M1) of a swine influenza virus (H1N1) causing chronic respiratory disease in pigs. Virus Genes 2000, 21, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Leser, G.P.; Jackson, D.; Lamb, R.A. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J. Virol. 2008, 82, 10059–10070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braza, F.; Brouard, S.; Chadban, S.; Goldstein, D.R. Role of TLRs and DAMPs in allograft inflammation and transplant outcomes. Nat. Rev. Nephrol. 2016, 12, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Mishina, M.; Ranjan, P.; De La Cruz, J.A.; Kim, J.H.; Garten, R.; Kumar, A.; Garcia-Sastre, A.; Katz, J.M.; Gangappa, S.; et al. A Newly Emerged Swine-Origin Influenza A(H3N2) Variant Dampens Host Antiviral Immunity but Induces Potent Inflammasome Activation. J. Infect. Dis. 2015, 212, 1923–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Zhu, Y.; Lin, X.; Ren, C.; Zhao, J.; Wang, F.; Gao, X.; Xiao, R.; Zhao, L.; Chen, H.; et al. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy 2019, 15, 1163–1181. [Google Scholar] [CrossRef] [PubMed]
- van Wielink, R.; Harmsen, M.M.; Martens, D.E.; Peeters, B.P.; Wijffels, R.H.; Moormann, R.J. Mutations in the M-gene segment can substantially increase replication efficiency of NS1 deletion influenza A virus in MDCK cells. J. Virol. 2012, 86, 12341–12350. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primer Sequences |
|---|---|
| CA09 M1 + H3N2 M2 inner-F | 5′-CTATCAGAAACGAATGGGGGTGCAGATGCAACG-3′ |
| CA09 M1 + H3N2 M2 inner-R | 5′-TCACTTGAATCGTTGCATCTGCACCCC-3′ |
| H3N2 M1 + CA09 M2 inner-F | 5′-ACCTACCAGAAGCGAATGGGAGTG-3′ |
| H3N2 M1 + CA09 M2 inner-R | 5′-TCACTTGAATCGCTGCATCTGCACTC-3′ |
| IL-1β | F: 5′-CACCTGGTACATCAGCACCTCAC-3′ |
| R: 5′-CATCAGAAACAGTCCAGCCCATAC-3′ | |
| IL-10 | F: 5′-GGTTGCCAAGCCTTATCGGA-3′ |
| R: 5′-ACCTGCTCCACTGCCTTGCT-3′ | |
| IFN-β | F: 5′-AAGAGTTACACTGCCTTTGCCATC-3′ |
| R: 5′-CACTGTCTGCTGGTGGAGTTCATC-3′ | |
| TNF-α | F: 5′-CGATGAGGTCAATCTGCCCA-3′ |
| R: 5′-CCAGGTCACTGTCCCAGCATC-3′ | |
| GAPDH | F: 5′-CATCACTGCCACCCAGAAGACTG-3′ |
| R: 5′-ATGCCAGTGAGCTTCCCGTTCAG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, L.; Xu, G.; Xin, L.; Wang, Z.; Wu, R.; Wu, M.; Cheng, Y.; Wang, H.; Yan, Y.; Ma, J.; et al. Eurasian Avian-like M1 Plays More Important Role than M2 in Pathogenicity of 2009 Pandemic H1N1 Influenza Virus in Mice. Viruses 2021, 13, 2335. https://doi.org/10.3390/v13122335
Xie L, Xu G, Xin L, Wang Z, Wu R, Wu M, Cheng Y, Wang H, Yan Y, Ma J, et al. Eurasian Avian-like M1 Plays More Important Role than M2 in Pathogenicity of 2009 Pandemic H1N1 Influenza Virus in Mice. Viruses. 2021; 13(12):2335. https://doi.org/10.3390/v13122335
Chicago/Turabian StyleXie, Lixiang, Guanlong Xu, Lingxiang Xin, Zhaofei Wang, Rujuan Wu, Mingqing Wu, Yuqiang Cheng, Hengan Wang, Yaxian Yan, Jingjiao Ma, and et al. 2021. "Eurasian Avian-like M1 Plays More Important Role than M2 in Pathogenicity of 2009 Pandemic H1N1 Influenza Virus in Mice" Viruses 13, no. 12: 2335. https://doi.org/10.3390/v13122335
APA StyleXie, L., Xu, G., Xin, L., Wang, Z., Wu, R., Wu, M., Cheng, Y., Wang, H., Yan, Y., Ma, J., & Sun, J. (2021). Eurasian Avian-like M1 Plays More Important Role than M2 in Pathogenicity of 2009 Pandemic H1N1 Influenza Virus in Mice. Viruses, 13(12), 2335. https://doi.org/10.3390/v13122335