
WGS- versus ORF5-Based Typing of PRRSV: A Belgian Case Study

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. RNA Extraction and DNase Treatment
2.3. cDNA Synthesis
2.4. High-Throughput Sequencing
2.5. Datasets
2.6. Distribution of Insertions and Deletions
2.7. Phylogenetic Analysis
2.8. Cluster Analysis
2.9. Recombination Analysis
3. Results
3.1. Whole Genome Sequencing Analysis
3.2. Insertions and Deletions
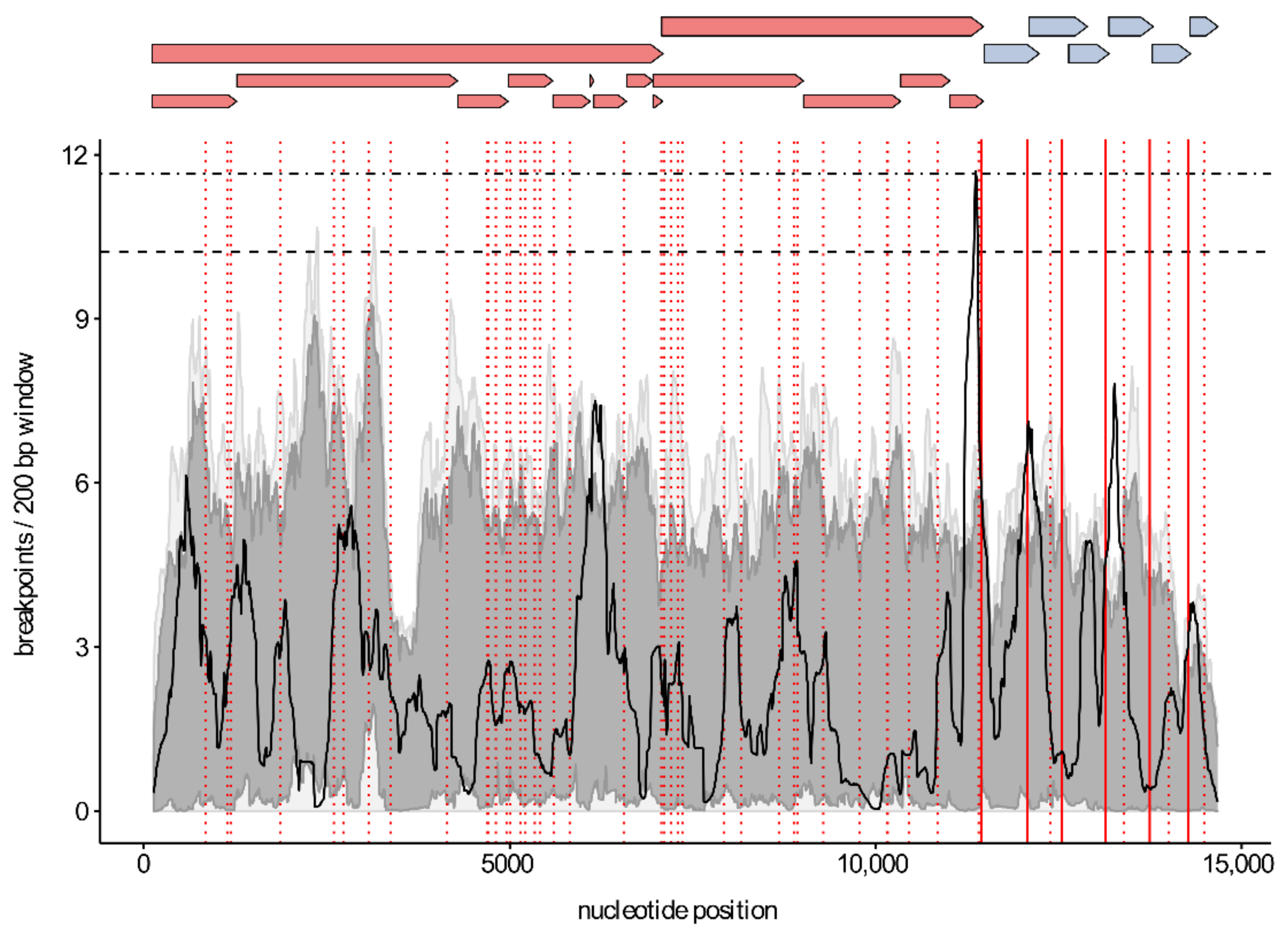
3.3. Recombination Analysis
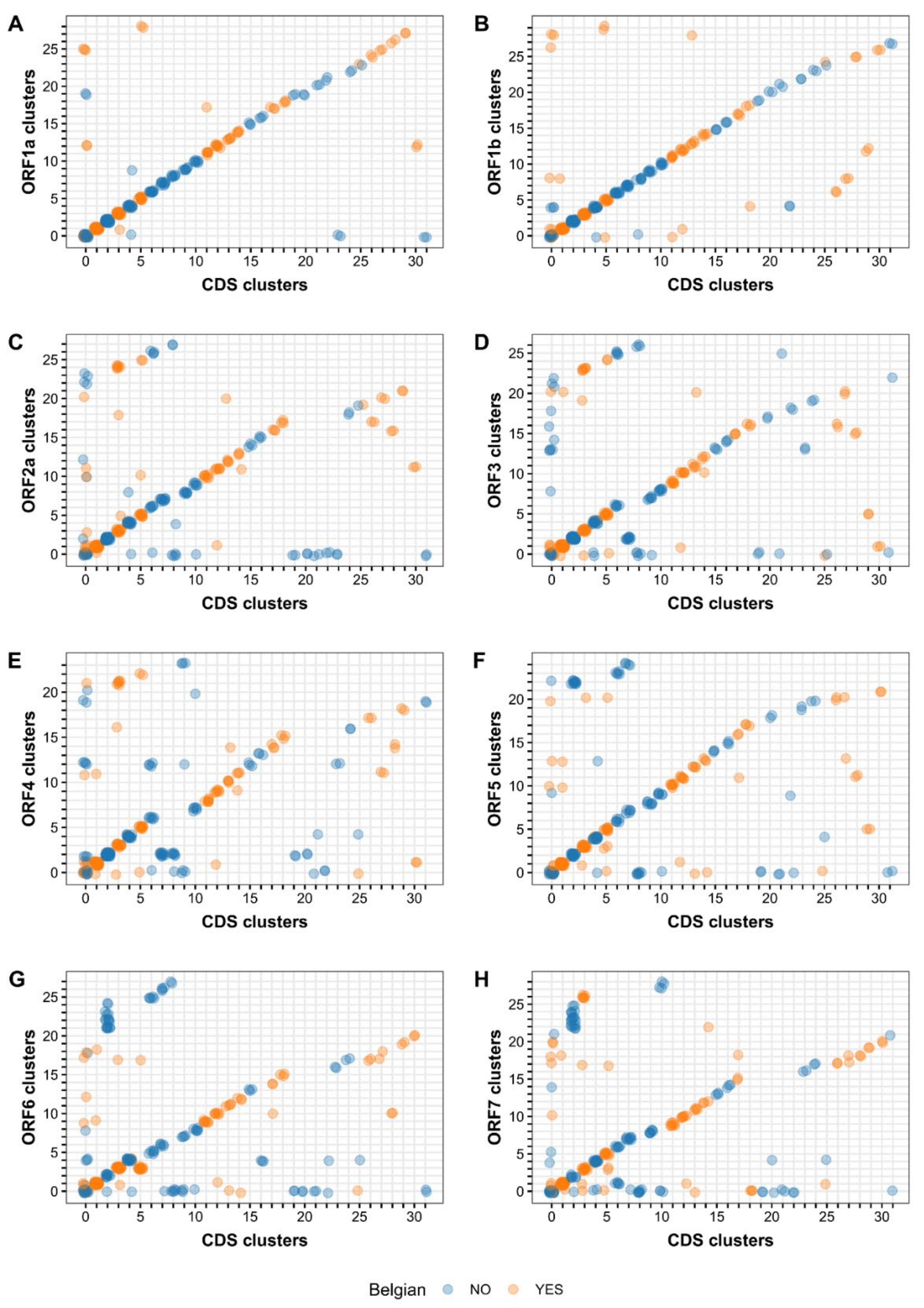
3.4. Phylogenetic Analysis of Separate versus Concatenated ORFs
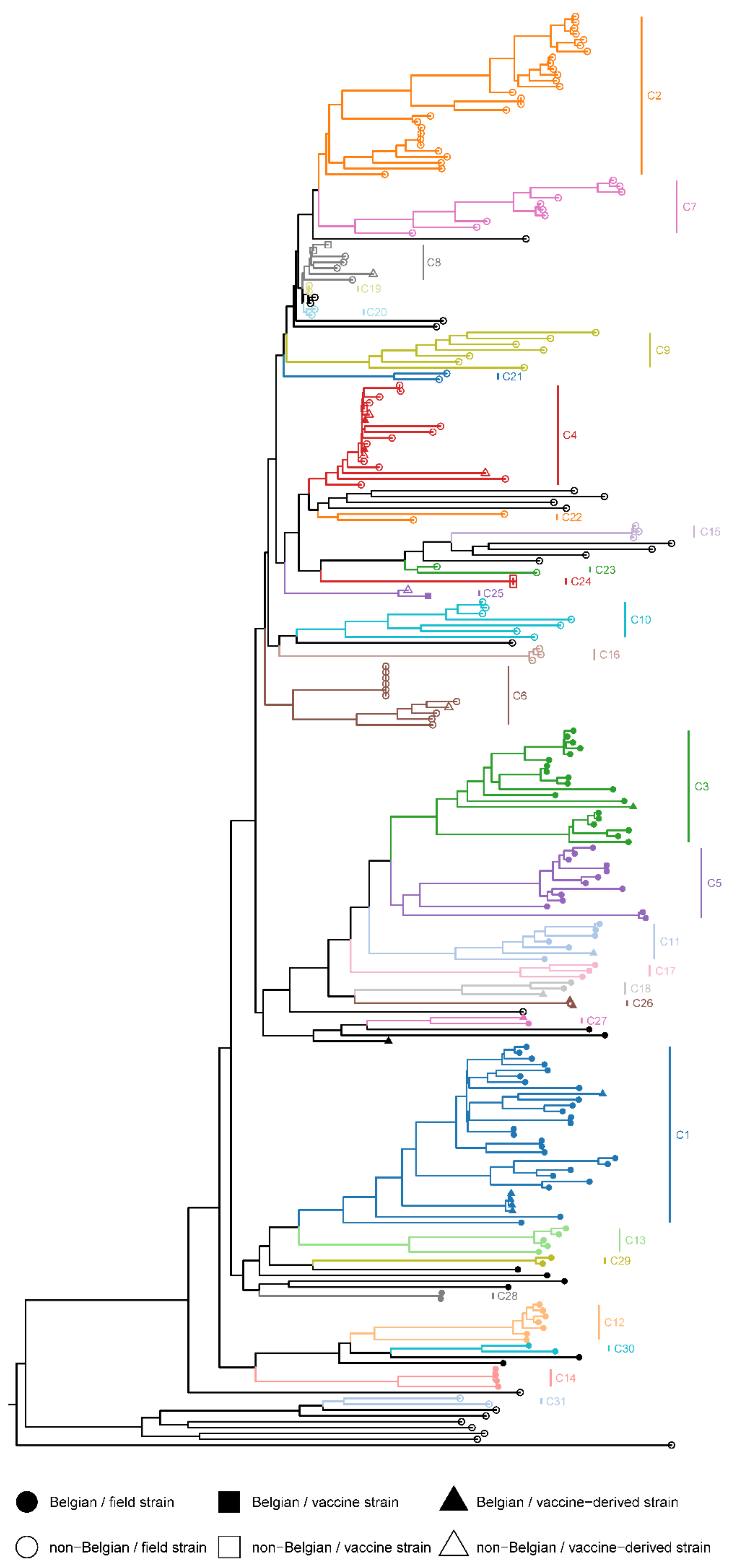
3.5. Phylogenetic Cluster Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zimmerman, J.J.; Dee, S.A.; Holtkamp, D.J.; Murtaugh, M.P.; Stadejek, T.; Stevenson, G.W.; Torremorell, M.; Yang, H.; Zhang, J. Porcine Reproductive and Respiratory Syndrome Viruses (Porcine Arteriviruses). In Diseases of Swine; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2019; pp. 685–708. ISBN 978-1-119-35092-7. [Google Scholar]
- Forsberg, R. Divergence Time of Porcine Reproductive and Respiratory Syndrome Virus Subtypes. Mol. Biol. Evol. 2005, 22, 2131–2134. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Lauck, M.; Bailey, A.L.; Shchetinin, A.M.; Vishnevskaya, T.V.; Bào, Y.; Ng, T.F.F.; LeBreton, M.; Schneider, B.S.; Gillis, A.; et al. Reorganization and Expansion of the Nidoviral Family Arteriviridae. Arch. Virol. 2016, 161, 755–768. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; Junglen, S.; et al. Changes to Virus Taxonomy and the International Code of Virus Classification and Nomenclature Ratified by the International Committee on Taxonomy of Viruses. Arch. Virol. 2019, 164, 2417–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappes, M.A.; Faaberg, K.S. PRRSV Structure, Replication and Recombination: Origin of Phenotype and Genotype Diversity. Virology 2015, 479–480, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, A.O.; Spaan, W.J.M.; Snijder, E.J.Y. Nidovirus Transcription: How to Make Sense? J. Gen. Virol. 2006, 87, 1403–1421. [Google Scholar] [CrossRef] [PubMed]
- Di, H.; McIntyre, A.A.; Brinton, M.A. New Insights about the Regulation of Nidovirus Subgenomic MRNA Synthesis. Virology 2018, 517, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Renukaradhya, G.J.; Meng, X.-J.; Calvert, J.G.; Roof, M.; Lager, K.M. Live Porcine Reproductive and Respiratory Syndrome Virus Vaccines: Current Status and Future Direction. Vaccine 2015, 33, 4069–4080. [Google Scholar] [CrossRef]
- Nan, Y.; Wu, C.; Gu, G.; Sun, W.; Zhang, Y.-J.; Zhou, E.-M. Improved Vaccine against PRRSV: Current Progress and Future Perspective. Front. Microbiol. 2017, 8, 1635. [Google Scholar] [CrossRef] [PubMed]
- Montaner-Tarbes, S.; Del Portillo, H.A.; Montoya, M.; Fraile, L. Key Gaps in the Knowledge of the Porcine Respiratory Reproductive Syndrome Virus (PRRSV). Front. Vet. Sci. 2019, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadejek, T.; Oleksiewicz, M.B.; Potapchuk, D.; Podgórska, K. Porcine Reproductive and Respiratory Syndrome Virus Strains of Exceptional Diversity in Eastern Europe Support the Definition of New Genetic Subtypes. J. Gen. Virol. 2006, 87, 1835–1841. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lam, T.T.-Y.; Hon, C.-C.; Hui, R.K.-H.; Faaberg, K.S.; Wennblom, T.; Murtaugh, M.P.; Stadejek, T.; Leung, F.C.-C. Molecular Epidemiology of PRRSV: A Phylogenetic Perspective. Virus Res. 2010, 154, 7–17. [Google Scholar] [CrossRef]
- Lambert, M.-È.; Arsenault, J.; Audet, P.; Delisle, B.; D’Allaire, S. Evaluating an Automated Clustering Approach in a Perspective of Ongoing Surveillance of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Field Strains. Infect. Genet. Evol. 2019, 73, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Stadejek, T.; Oleksiewicz, M.B.; Scherbakov, A.V.; Timina, A.M.; Krabbe, J.S.; Chabros, K.; Potapchuk, D. Definition of Subtypes in the European Genotype of Porcine Reproductive and Respiratory Syndrome Virus: Nucleocapsid Characteristics and Geographical Distribution in Europe. Arch. Virol. 2008, 153, 1479–1488. [Google Scholar] [CrossRef]
- Murtaugh, M.P.; Stadejek, T.; Abrahante, J.E.; Lam, T.T.Y.; Leung, F.C.-C. The Ever-Expanding Diversity of Porcine Reproductive and Respiratory Syndrome Virus. Virus Res. 2010, 154, 18–30. [Google Scholar] [CrossRef]
- Stadejek, T.; Stankevicius, A.; Murtaugh, M.P.; Oleksiewicz, M.B. Molecular Evolution of PRRSV in Europe: Current State of Play. Vet. Microbiol. 2013, 165, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Kvisgaard, L.K.; Hjulsager, C.K.; Fahnøe, U.; Breum, S.Ø.; Ait-Ali, T.; Larsen, L.E. A Fast and Robust Method for Full Genome Sequencing of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Type 1 and Type 2. J. Virol. Methods 2013, 193, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zheng, Y.; Xia, X.-Q.; Chen, Q.; Bade, S.A.; Yoon, K.-J.; Harmon, K.M.; Gauger, P.C.; Main, R.G.; Li, G. High-Throughput Whole Genome Sequencing of Porcine Reproductive and Respiratory Syndrome Virus from Cell Culture Materials and Clinical Specimens Using Next-Generation Sequencing Technology. J. Vet. Diagn. Investig. 2017, 29, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Yan, Y.; Shi, M.; Liu, H.-Z.; Zhang, H.-L.; Yang, Y.-B.; Huang, X.-Y.; Gauger, P.C.; Zhang, J.; Zhang, Y.-H.; et al. Phylogenetics, Genomic Recombination, and NSP2 Polymorphic Patterns of Porcine Reproductive and Respiratory Syndrome Virus in China and the United States in 2014–2018. J. Virol. 2020, 94, e01813–e01819. [Google Scholar] [CrossRef] [PubMed]
- Rojo-Gimeno, C.; Dewulf, J.; Maes, D.; Wauters, E. A Systemic Integrative Framework to Describe Comprehensively a Swine Health System, Flanders as an Example. Prev. Vet. Med. 2018, 154, 30–46. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinform. Oxf. Engl. 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. In Proceedings of the German Conference on Bioinformatics, Hannover, Germany, 4–6 October 1999. [Google Scholar]
- Hunt, M.; Gall, A.; Ong, S.H.; Brener, J.; Ferns, B.; Goulder, P.; Nastouli, E.; Keane, J.A.; Kellam, P.; Otto, T.D. IVA: Accurate de Novo Assembly of RNA Virus Genomes. Bioinform. Oxf. Engl. 2015, 31, 2374–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Schäffer, A.A.; Hatcher, E.L.; Yankie, L.; Shonkwiler, L.; Brister, J.R.; Karsch-Mizrachi, I.; Nawrocki, E.P. VADR: Validation and Annotation of Virus Sequence Submissions to GenBank. BMC Bioinform. 2020, 21, 211. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menardo, F.; Loiseau, C.; Brites, D.; Coscolla, M.; Gygli, S.M.; Rutaihwa, L.K.; Trauner, A.; Beisel, C.; Borrell, S.; Gagneux, S. Treemmer: A Tool to Reduce Large Phylogenetic Datasets with Minimal Loss of Diversity. BMC Bioinform. 2018, 19, 164. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.S. Using DECIPHER v2.0 to Analyze Big Biological Sequence Data in R. R J. 2016, 8, 352. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Naser-Khdour, S.; Minh, B.Q.; Zhang, W.; Stone, E.A.; Lanfear, R. The Prevalence and Impact of Model Violations in Phylogenetic Analysis. Genome Biol. Evol. 2019, 11, 3341–3352. [Google Scholar] [CrossRef] [Green Version]
- Chernomor, O.; von Haeseler, A.; Minh, B.Q. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. Model Finder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strimmer, K.; Haeseler, A. von Likelihood-Mapping: A Simple Method to Visualize Phylogenetic Content of a Sequence Alignment. Proc. Natl. Acad. Sci. USA 1997, 94, 6815–6819. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Prosperi, M.C.F.; Ciccozzi, M.; Fanti, I.; Saladini, F.; Pecorari, M.; Borghi, V.; Di Giambenedetto, S.; Bruzzone, B.; Capetti, A.; Vivarelli, A.; et al. A Novel Methodology for Large-Scale Phylogeny Partition. Nat. Commun. 2011, 2, 321. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation. 2020. Available online: https://CRAN.R-project.org/package=dplyr (accessed on 1 April 2021).
- Pennell, M.W.; Eastman, J.M.; Slater, G.J.; Brown, J.W.; Uyeda, J.C.; FitzJohn, R.G.; Alfaro, M.E.; Harmon, L.J. Geiger v2.0: An Expanded Suite of Methods for Fitting Macroevolutionary Models to Phylogenetic Trees. Bioinformatics 2014, 30, 2216–2218. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Wang, L.-G.; Lam, T.T.-Y.; Xu, S.; Dai, Z.; Zhou, L.; Feng, T.; Guo, P.; Dunn, C.W.; Jones, B.R.; Bradley, T.; et al. Treeio: An R Package for Phylogenetic Tree Input and Output with Richly Annotated and Associated Data. Mol. Biol. Evol. 2020, 37, 599–603. [Google Scholar] [CrossRef]
- Dragulescu, A.; Arendt, C. Xlsx: Read, Write, Format Excel 2007 and Excel 97/2000/XP/2003 Files. 2020. Available online: https://CRAN.R-project.org/package=xlsx (accessed on 1 April 2021).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Vinh, N.X.; Epps, J.; Bailey, J. Information Theoretic Measures for Clusterings Comparison: Is a Correction for Chance Necessary? In Proceedings of the 26th Annual International Conference on Machine Learning, New York, NY, USA, 14 June 2009; Association for Computing Machinery: New York, NY, USA, 2009; pp. 1073–1080. [Google Scholar]
- Bagga, A.; Baldwin, B. Entity-Based Cross-Document Coreferencing Using the Vector Space Model. In Proceedings of the 36th Annual Meeting of the Association for Computational Linguistics and 17th International Conference on Computational Linguistics, Montreal, QC, Canada, 10 August 1998; Association for Computational Linguistics: Stroudsburg, PA, USA, 1998; Volume 1, pp. 79–85. [Google Scholar]
- Chiquet, J.; Rigaill, G.; Sundqvist, M. Aricode: Efficient Computations of Standard Clustering Comparison Measures. 2020. Available online: https://CRAN.R-project.org/package=aricode (accessed on 1 April 2021).
- Zhang, L. DPBBM: Dirichlet Process Beta-Binomial Mixture. 2016. Available online: https://CRAN.R-project.org/package=DPBBM (accessed on 1 April 2021).
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A Computer Program for Analysing Recombination in, and Removing Signals of Recombination from, Nucleotide Sequence Datasets. Virus Evol. 2020, 7, veaa087. [Google Scholar] [CrossRef]
- Revell, L.J. Phytools: An R Package for Phylogenetic Comparative Biology (and Other Things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Tan, S.; Dvorak, C.M.T.; Murtaugh, M.P. Rapid, Unbiased PRRSV Strain Detection Using MinION Direct RNA Sequencing and Bioinformatics Tools. Viruses 2019, 11, 1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsberg, R.; Storgaard, T.; Nielsen, H.S.; Oleksiewicz, M.B.; Cordioli, P.; Sala, G.; Hein, J.; Bøtner, A. The Genetic Diversity of European Type PRRSV Is Similar to That of the North American Type but Is Geographically Skewed within Europe. Virology 2002, 299, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Valls, G.E.; Kvisgaard, L.K.; Tello, M.; Darwich, L.; Cortey, M.; Burgara-Estrella, A.J.; Hernández, J.; Larsen, L.E.; Mateu, E. Analysis of ORF5 and Full-Length Genome Sequences of Porcine Reproductive and Respiratory Syndrome Virus Isolates of Genotypes 1 and 2 Retrieved Worldwide Provides Evidence That Recombination Is a Common Phenomenon and May Produce Mosaic Isolates. J. Virol. 2014, 88, 3170–3181. [Google Scholar] [CrossRef] [Green Version]
- Darwich, L.; Gimeno, M.; Sibila, M.; Diaz, I.; de la Torre, E.; Dotti, S.; Kuzemtseva, L.; Martin, M.; Pujols, J.; Mateu, E. Genetic and Immunobiological Diversities of Porcine Reproductive and Respiratory Syndrome Genotype I Strains. Vet. Microbiol. 2011, 150, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Kvisgaard, L.K.; Hjulsager, C.K.; Kristensen, C.S.; Lauritsen, K.T.; Larsen, L.E. Genetic and Antigenic Characterization of Complete Genomes of Type 1 Porcine Reproductive and Respiratory Syndrome Viruses (PRRSV) Isolated in Denmark over a Period of 10 Years. Virus Res. 2013, 178, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, R.; Oleksiewicz, M.B.; Krabbe Petersen, A.-M.; Hein, J.; Bøtner, A.; Storgaard, T. A Molecular Clock Dates the Common Ancestor of European-Type Porcine Reproductive and Respiratory Syndrome Virus at More Than 10 Years before the Emergence of Disease. Virology 2001, 289, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Oleksiewicz, M.B.; Bøtner, A.; Toft, P.; Grubbe, T.; Nielsen, J.; Kamstrup, S.; Storgaard, T. Emergence of Porcine Reproductive and Respiratory Syndrome Virus Deletion Mutants: Correlation with the Porcine Antibody Response to a Hypervariable Site in the ORF 3 Structural Glycoprotein. Virology 2000, 267, 135–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dortmans, J.C.F.M.; Buter, G.J.; Dijkman, R.; Houben, M.; Duinhof, T.F. Molecular Characterization of Type 1 Porcine Reproductive and Respiratory Syndrome Viruses (PRRSV) Isolated in the Netherlands from 2014 to 2016. PLoS ONE 2019, 14, e0218481. [Google Scholar] [CrossRef] [PubMed]
- Bøtner, A.; Strandbygaard, B.; Sørensen, K.J.; Have, P.; Madsen, K.G.; Madsen, E.S.; Alexandersen, S. Appearance of Acute PRRS-like Symptoms in Sow Herds after Vaccination with a Modified Live PRRS Vaccine. Vet. Rec. 1997, 141, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Grosse Beilage, E.; Nathues, H.; Meemken, D.; Harder, T.C.; Doherr, M.G.; Grotha, I.; Greiser-Wilke, I. Frequency of PRRS Live Vaccine Virus (European and North American Genotype) in Vaccinated and Non-Vaccinated Pigs Submitted for Respiratory Tract Diagnostics in North-Western Germany. Prev. Vet. Med. 2009, 92, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kiss, I.; Sámi, L.; Kecskeméti, S.; Hanada, K. Genetic Variation of the Prevailing Porcine Respiratory and Reproductive Syndrome Viruses Occurring on a Pig Farm upon Vaccination. Arch. Virol. 2006, 151, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Eclercy, J.; Renson, P.; Lebret, A.; Hirchaud, E.; Normand, V.; Andraud, M.; Paboeuf, F.; Blanchard, Y.; Rose, N.; Bourry, O. A Field Recombinant Strain Derived from Two Type 1 Porcine Reproductive and Respiratory Syndrome Virus (PRRSV-1) Modified Live Vaccines Shows Increased Viremia and Transmission in SPF Pigs. Viruses 2019, 11, 296. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.H.; Kim, H.; Kim, J.; Lee, H.-K.; Park, B.; Kim, H. Complete Genome Sequences of Porcine Reproductive and Respiratory Syndrome Viruses: Perspectives on Their Temporal and Spatial Dynamics. Mol. Biol. Rep. 2013, 40, 6843–6853. [Google Scholar] [CrossRef] [PubMed]
- Kvisgaard, L.K.; Kristensen, C.S.; Ryt-Hansen, P.; Pedersen, K.; Stadejek, T.; Trebbien, R.; Andresen, L.O.; Larsen, L.E. A Recombination between Two Type 1 Porcine Reproductive and Respiratory Syndrome Virus (PRRSV-1) Vaccine Strains Has Caused Severe Outbreaks in Danish Pigs. Transbound. Emerg. Dis. 2020, 67, 1786–1796. [Google Scholar] [CrossRef] [Green Version]
- Renson, P.; Touzain, F.; Lebret, A.; Le Dimna, M.; Quenault, H.; Normand, V.; Claude, J.-B.; Pez, F.; Rose, N.; Blanchard, Y.; et al. Complete Genome Sequence of a Recombinant Porcine Reproductive and Respiratory Syndrome Virus Strain from Two Genotype 1 Modified Live Virus Vaccine Strains. Genome Announc. 2017, 5, e00454-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Liu, Q.; Qiao, M.; Deng, X.; Chen, X.; Sun, M. Whole Genome Characterization of a Novel Porcine Reproductive and Respiratory Syndrome Virus 1 Isolate: Genetic Evidence for Recombination between Amervac Vaccine and Circulating Strains in Mainland China. Infect. Genet. Evol. 2017, 54, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Marton, S.; Szalay, D.; Kecskeméti, S.; Forró, B.; Olasz, F.; Zádori, Z.; Szabó, I.; Molnár, T.; Bányai, K.; Bálint, Á. Coding-Complete Sequence of a Vaccine-Derived Recombinant Porcine Reproductive and Respiratory Syndrome Virus Strain Isolated in Hungary. Arch. Virol. 2019, 164, 2605–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzo, G.; Cecchinato, M.; Martini, M.; Ceglie, L.; Gigli, A.; Drigo, M. Observation of High Recombination Occurrence of Porcine Reproductive and Respiratory Syndrome Virus in Field Condition. Virus Res. 2014, 194, 159–166. [Google Scholar] [CrossRef]
- Lukashev, A.N. Role of Recombination in Evolution of Enteroviruses. Rev. Med. Virol. 2005, 15, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Heath, L.; Walt, E. van der Varsani, A.; Martin, D.P. Recombination Patterns in Aphthoviruses Mirror Those Found in Other Picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, P. Recombination and Selection in the Evolution of Picornaviruses and Other Mammalian Positive-Stranded RNA Viruses. J. Virol. 2006, 80, 11124–11140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, P.; Welch, J. Frequency and Dynamics of Recombination within Different Species of Human Enteroviruses. J. Virol. 2006, 80, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig-Begall, L.F.; Mauroy, A.; Thiry, E. Norovirus Recombinants: Recurrent in the Field, Recalcitrant in the Lab—A Scoping Review of Recombination and Recombinant Types of Noroviruses. J. Gen. Virol. 2018, 99, 970–988. [Google Scholar] [CrossRef] [PubMed]
- Mahar, J.E.; Jenckel, M.; Huang, N.; Smertina, E.; Holmes, E.C.; Strive, T.; Hall, R.N. Frequent Intergenotypic Recombination between the Non-Structural and Structural Genes Is a Major Driver of Epidemiological Fitness in Caliciviruses. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Lett, J.-M.; Varsani, A.; Martin, D.P. Widely Conserved Recombination Patterns among Single-Stranded DNA Viruses. J. Virol. 2009, 83, 2697–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Negroni, M.; Robertson, D.L. The Distribution of HIV-1 Recombination Breakpoints. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2007, 7, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why Do RNA Viruses Recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.; Evans, D.J.Y. 2018 Mechanisms and Consequences of Positive-Strand RNA Virus Recombination. J. Gen. Virol. 2018, 99, 1345–1356. [Google Scholar] [CrossRef]
- Martin, D.P.; van der Walt, E.; Posada, D.; Rybicki, E.P. The Evolutionary Value of Recombination Is Constrained by Genome Modularity. PLoS Genet. 2005, 1, e51. [Google Scholar] [CrossRef]
- Molenkamp, R.; Greve, S.; Spaan, W.J.; Snijder, E.J. Efficient Homologous RNA Recombination and Requirement for an Open Reading Frame during Replication of Equine Arteritis Virus Defective Interfering RNAs. J. Virol. 2000, 74, 9062–9070. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yan, W.; Hall, A.B.; Jiang, X. Characterizing Transcriptional Regulatory Sequences in Coronaviruses and Their Role in Recombination. Mol. Biol. Evol. 2021, 38, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Bian, T.; Sun, Y.; Hao, M.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. A Recombinant Type 2 Porcine Reproductive and Respiratory Syndrome Virus between NADC30-like and a MLV-like: Genetic Characterization and Pathogenicity for Piglets. Infect. Genet. Evol. 2017, 54, 279–286. [Google Scholar] [CrossRef]
- Lu, W.H.; Tun, H.M.; Sun, B.L.; Mo, J.; Zhou, Q.F.; Deng, Y.X.; Xie, Q.M.; Bi, Y.Z.; Leung, F.C.-C.; Ma, J.Y. Re-Emerging of Porcine Respiratory and Reproductive Syndrome Virus (Lineage 3) and Increased Pathogenicity after Genomic Recombination with Vaccine Variant. Vet. Microbiol. 2015, 175, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, C.S.; Christiansen, M.G.; Pedersen, K.; Larsen, L.E. Production Losses Five Months after Outbreak with a Recombinant of Two PRRSV Vaccine Strains in 13 Danish Sow Herds. Porc. Health Manag. 2020, 6, 26. [Google Scholar] [CrossRef]
- Yount, B.; Roberts, R.S.; Lindesmith, L.; Baric, R.S. Rewiring the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Transcription Circuit: Engineering a Recombination-Resistant Genome. Proc. Natl. Acad. Sci. USA 2006, 103, 12546–12551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, R.L.; Deming, D.J.; Deming, M.E.; Yount, B.L.; Baric, R.S. Evaluation of a Recombination-Resistant Coronavirus as a Broadly Applicable, Rapidly Implementable Vaccine Platform. Commun. Biol. 2018, 1, 1–10. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORF | Protein | Belgian Dataset Internal/Terminal InDels | Complete Dataset Internal/Terminal InDels |
|---|---|---|---|
| ORF1ab | NSP1 | 0/0 | 6/0 |
| ORF1ab | NSP2 | 50/0 | 122/0 |
| ORF1ab | NSP3 | 0/0 | 0/0 |
| ORF1ab | NSP4 | 0/0 | 0/0 |
| ORF1ab | NSP5 | 0/0 | 0/0 |
| ORF1ab | NSP6 | 0/0 | 0/0 |
| ORF1ab | NSP7α | 0/0 | 0/0 |
| ORF1ab | NSP7β | 0/0 | 1/0 |
| ORF1ab | NSP8 | 0/0 | 0/0 |
| ORF1ab | NSP9 | 0/0 | 0/0 |
| ORF1ab | NSP10 | 0/0 | 1/0 |
| ORF1ab | NSP11 | 0/0 | 0/0 |
| ORF1ab | NSP12 | 1/5 | 1/7 |
| ORF2 | GP2 | 0/1 | 1/1 |
| ORF3 | GP3 | 9/12 | 18/15 |
| ORF4 | GP4 | 11/0 | 22/0 |
| ORF5 | GP5 | 0/0 | 0/0 |
| ORF6 | M | 1/0 | 1/0 |
| ORF7 | N | 0/0 | 2/2 |
| Event | Recombinant | Major Parent | Minor Parent | Start/Stop Position | Region Exchanged |
|---|---|---|---|---|---|
| 7 | MZ417409 | MZ417402 | KT988004 | 11,927/13,412 | ORF2-ORF5 |
| 15 | MZ417409 | MZ417402 | KT988004 | 13,413/14,733 | ORF5-ORF7 |
| 5 | MZ417426 | MZ417421 | DD093450 | 6049/9047 | ORF1a-ORF1b |
| 36 | MZ417449 | MZ417446 | KT988004 | 8104/8543 | ORF1b |
| 9 a | MZ417459 | MZ417446 | DD093450 | 6514/8680 | ORF1a-ORF1b |
| 13 | MZ417463 | MZ417462 | KT988004 | 13,157/14,587 | ORF5-ORF7 |
| 2 | MZ417464 | MZ417463 | DD093450 | 2721/10,884 | ORF1a-ORF1b |
| 13 | MZ417464 | MZ417462 | KT988004 | 13,232/14,587 | ORF5-ORF7 |
| 3 | MZ417469 | MZ417467 | GU067771 | 6271/10,979 | ORF1a-ORF1b |
| 51 | MZ417469 | MZ417467 | GU067771 | 1/542 | ORF1a |
| 20 b | MZ417496 | MZ417417 | KT988004 | 13,366/14,695 | ORF5-ORF7 |
| Dataset | Length Alignment | % Fully Resolved Quartets | % Partly Resolved Quartets | % Unresolved Quartets |
|---|---|---|---|---|
| CDS | 15,591 | 97.65 | 2.24 | 0.11 |
| ORF1a | 7506 | 94.85 | 4.53 | 0.62 |
| ORF1b | 4458 | 89.26 | 8.81 | 1.93 |
| ORF2a | 747 | 66.06 | 15.89 | 18.05 |
| ORF3 | 810 | 74.79 | 15.28 | 9.92 |
| ORF4 | 555 | 66.94 | 19.58 | 13.48 |
| ORF5 | 603 | 82.62 | 9.20 | 8.18 |
| ORF6 | 519 | 75.04 | 13.67 | 11.29 |
| ORF7 | 393 | 61.40 | 13.85 | 24.75 |
| Dataset | AMI | F(b3) |
|---|---|---|
| CDS/ORF1a | 0.93 | 0.90 |
| CDS/ORF1b | 0.90 | 0.86 |
| CDS/ORF2a | 0.81 | 0.75 |
| CDS/ORF3 | 0.80 | 0.71 |
| CDS/ORF4 | 0.74 | 0.68 |
| CDS/ORF5 | 0.79 | 0.73 |
| CDS/ORF6 | 0.74 | 0.65 |
| CDS/ORF7 | 0.73 | 0.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vandenbussche, F.; Mathijs, E.; Tignon, M.; Vandersmissen, T.; Cay, A.B. WGS- versus ORF5-Based Typing of PRRSV: A Belgian Case Study. Viruses 2021, 13, 2419. https://doi.org/10.3390/v13122419
Vandenbussche F, Mathijs E, Tignon M, Vandersmissen T, Cay AB. WGS- versus ORF5-Based Typing of PRRSV: A Belgian Case Study. Viruses. 2021; 13(12):2419. https://doi.org/10.3390/v13122419
Chicago/Turabian StyleVandenbussche, Frank, Elisabeth Mathijs, Marylène Tignon, Tamara Vandersmissen, and Ann Brigitte Cay. 2021. "WGS- versus ORF5-Based Typing of PRRSV: A Belgian Case Study" Viruses 13, no. 12: 2419. https://doi.org/10.3390/v13122419
APA StyleVandenbussche, F., Mathijs, E., Tignon, M., Vandersmissen, T., & Cay, A. B. (2021). WGS- versus ORF5-Based Typing of PRRSV: A Belgian Case Study. Viruses, 13(12), 2419. https://doi.org/10.3390/v13122419