
Virome Characterization in Commercial Bovine Serum Batches—A Potentially Needed Testing Strategy for Biological Products


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples of the Commercial Bovine Serum Batches
2.2. Sample Preparation and Sequencing
2.3. Bioinformatic Analysis
3. Results
3.1. Overview
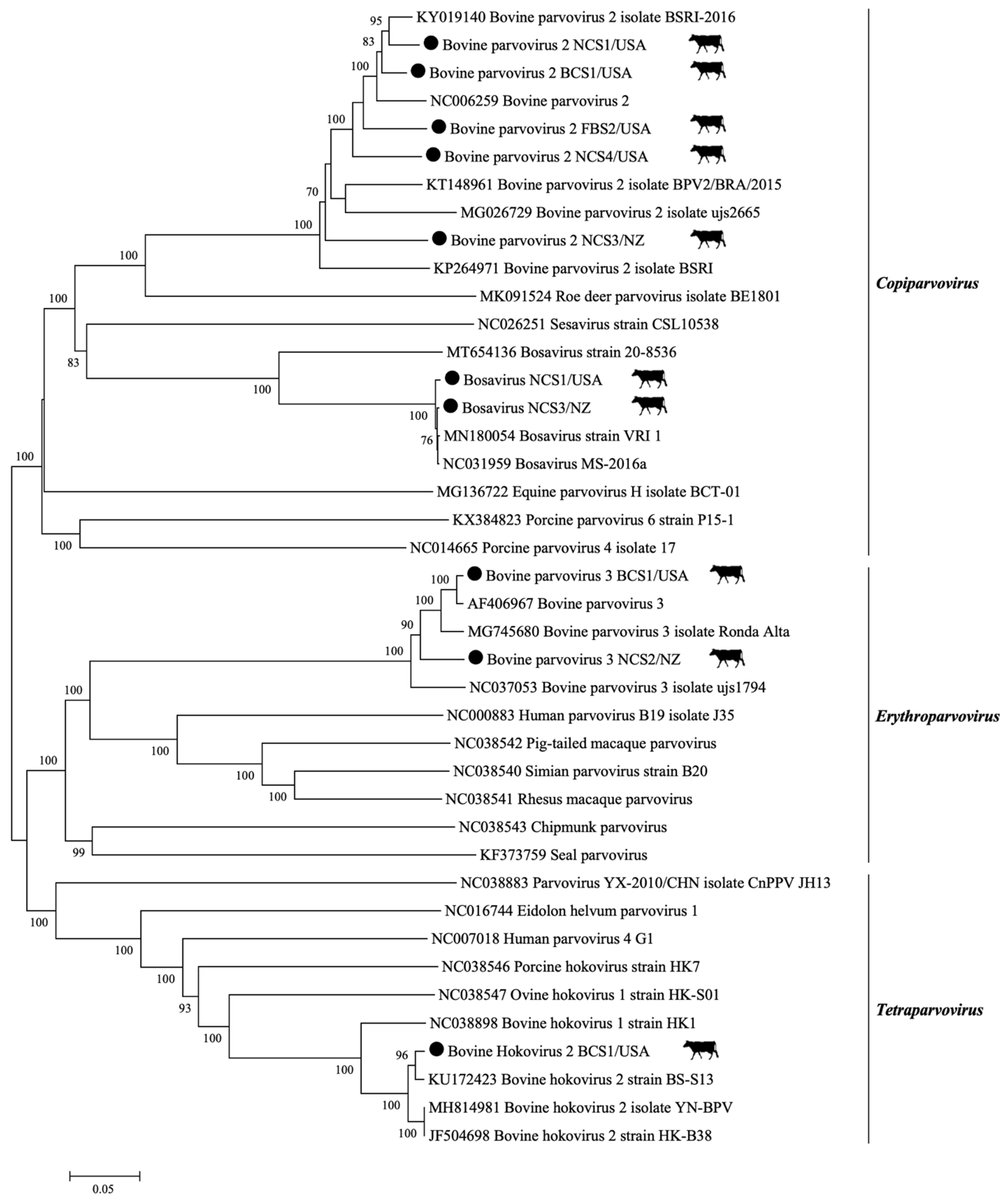
3.2. DNA Viral Families: Parvoviridae, Polyomaviridae, and Adenoviridae
3.3. RNA Viruses: Flaviviridae, Picornaviridae, Reoviridae, and Retroviridae
3.4. Circular Rep-Encoding Single-Stranded (CRESS) DNA Viruses-Genomoviridae, Circoviridae, and Smacoviridae
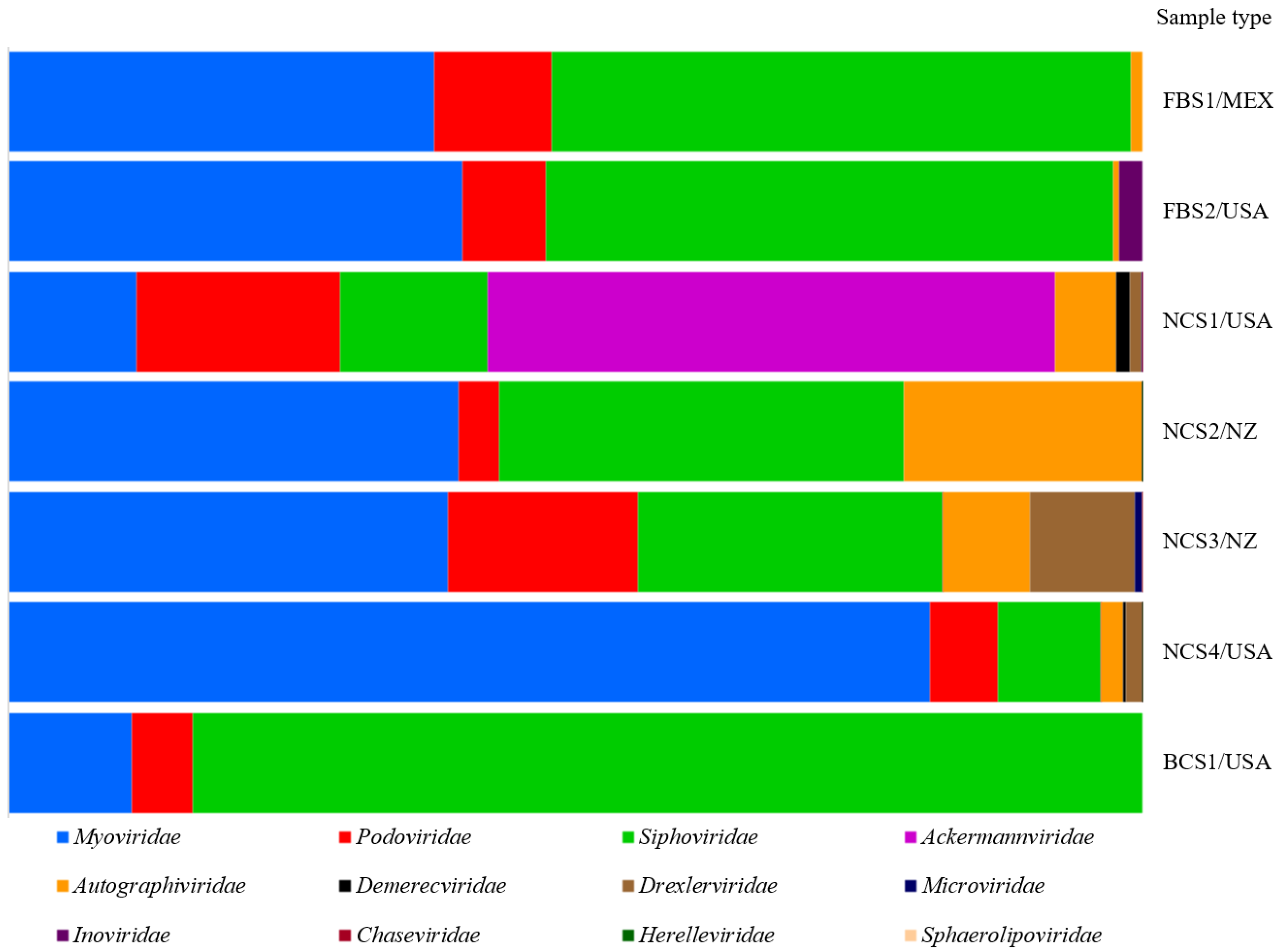
3.5. Bacteriophages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- van der Valk, J.; Bieback, K.; Buta, C.; Cochrane, B.; Dirks, W.G.; Fu, J.; Hickman, J.J.; Hohensee, C.; Kolar, R.; Liebsch, M.; et al. Fetal Bovine Serum (FBS): Past–Present–Future. ALTEX 2018, 35, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Sykes, J.E.; Rankin, S.C. Chapter 1—Isolation in Cell Culture. In Canine and Feline Infectious Diseases; Sykes, J.E., Ed.; Saunders: Saint Louis, MO, USA, 2014; pp. 2–9. ISBN 978-1-4377-0795-3. [Google Scholar]
- Stephens, N.; Di Silvio, L.; Dunsford, I.; Ellis, M.; Glencross, A.; Sexton, A. Bringing cultured meat to market: Technical, socio-political, and regulatory challenges in cellular agriculture. Trends Food Sci. Technol. 2018, 78, 155–166. [Google Scholar] [CrossRef]
- Verma, A.; Verma, M.; Singh, A. Animal tissue culture principles and applications. Anim. Biotechnol. 2020, 269–293. [Google Scholar] [CrossRef]
- Murray, J.; Versteegen, R. Fetal Bovine—Geographical Origin and International Trade. Bioprocess. J. 2019, 18. [Google Scholar] [CrossRef]
- Baylis, S.A.; Miskey, C.; Blümel, J.; Kaiser, M.; Kapusinszky, B.; Delwart, E. Identification of a novel bovine copiparvovirus in pooled fetal bovine serum. Virus Genes 2020, 56, 522–526. [Google Scholar] [CrossRef]
- Paim, W.P.; Maggioli, M.F.; Weber, M.N.; Rezabek, G.; Narayanan, S.; Ramachandran, A.; Canal, C.W.; Bauermann, F.V. Virome characterization in serum of healthy show pigs raised in Oklahoma demonstrated great diversity of ssDNA viruses. Virology 2021, 556, 87–95. [Google Scholar] [CrossRef]
- Toohey-Kurth, K.; Sibley, S.D.; Goldberg, T.L. Metagenomic assessment of adventitious viruses in commercial bovine sera. Biologicals 2017, 47, 64–68. [Google Scholar] [CrossRef]
- Sadeghi, M.; Kapusinszky, B.; Yugo, D.M.; Phan, T.G.; Deng, X.; Kanevsky, I.; Opriessnig, T.; Woolums, A.R.; Hurley, D.J.; Meng, X.J.; et al. Virome of US bovine calf serum. Biologicals 2017, 46, 64–67. [Google Scholar] [CrossRef] [Green Version]
- Cheval, J.; Sauvage, V.; Frangeul, L.; Dacheux, L.; Guigon, G.; Dumey, N.; Pariente, K.; Rousseaux, C.; Dorange, F.; Berthet, N.; et al. Evaluation of high-throughput sequencing for identifying known and unknown viruses in biological samples. J. Clin. Microbiol. 2011, 49, 3268–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnieur, L.; Cheval, J.; Gratigny, M.; Hébert, C.; Muth, E.; Dumarest, M.; Eloit, M. Unbiased analysis by high throughput sequencing of the viral diversity in fetal bovine serum and trypsin used in cell culture. Biologicals 2014, 42, 145–152. [Google Scholar] [CrossRef]
- Victoria, J.G.; Wang, C.; Jones, M.S.; Jaing, C.; McLoughlin, K.; Gardner, S.; Delwart, E.L. Viral nucleic acids in live-attenuated vaccines: Detection of minority variants and an adventitious virus. J. Virol. 2010, 84, 6033–6040. [Google Scholar] [CrossRef] [Green Version]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef]
- Sambrook, J.; Russel, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; 2344p. [Google Scholar]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Caesar, L.; Cibulski, S.P.; Canal, C.W.; Blochtein, B.; Sattler, A.; Haag, K.L. The virome of an endangered stingless bee suffering from annual mortality in Southern Brazil. J. Gen. Virol. 2019, 100, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Pecora, A.; Pérez López, J.; Jordán, M.J.; Franco, L.N.; Politzki, R.; Ruiz, V.; Alvarez, I. Analysis of irradiated Argentinean fetal bovine serum for adventitious agents. J. Vet. Diagn. Investig. 2020, 32, 892–897. [Google Scholar] [CrossRef]
- Gauvin, G.; Nims, R. Gamma-irradiation of serum for the inactivation of adventitious contaminants. PDA J. Pharm. Sci. Technol. 2010, 64, 432–435. [Google Scholar]
- Nims, R.W.; Gauvin, G.; Plavsic, M. Gamma irradiation of animal sera for inactivation of viruses and mollicutes—A review. Biologicals 2011, 39, 370–377. [Google Scholar] [CrossRef]
- House, C.; House, J.A.; Yedloutschnig, R.J. Inactivation of viral agents in bovine serum by gamma irradiation. Can. J. Microbiol. 1990, 36, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Plavsic, M.; Nims, R.; Wintgens, M.; Versteegen, R. Gamma irradiation of animal serum: Validation of efficacy for pathogen reduction and assessment of impacts on serum performance. Bioprocessing 2016, 15, 1538–8786. [Google Scholar] [CrossRef]
- Wang, H.; Li, S.; Mahmood, A.; Yang, S.; Wang, X.; Shen, Q.; Shan, T.; Deng, X.; Li, J.; Hua, X.; et al. Plasma virome of cattle from forest region revealed diverse small circular ssDNA viral genomes. Virol. J. 2018, 15, 11. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.N.; Cibulski, S.P.; Silveira, S.; Siqueira, F.M.; Mósena, A.C.S.; da Silva, M.S.; Olegário, J.C.; Varela, A.P.M.; Teixeira, T.F.; Bianchi, M.V.; et al. Evaluation of the serum virome in calves persistently infected with Pestivirus A, presenting or not presenting mucosal disease. Virus Genes 2018, 54, 768–778. [Google Scholar] [CrossRef]
- Binga, E.K.; Lasken, R.S.; Neufeld, J.D. Something from (almost) nothing: The impact of multiple displacement amplification on microbial ecology. ISME J. 2008, 2, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Kirikae, T.; Tamura, H.; Hashizume, M.; Kirikae, F.; Uemura, Y.; Tanaka, S.; Yokochi, T.; Nakano, M. Endotoxin contamination in fetal bovine serum and its influence on tumor necrosis factor production by macrophage-like cells J774.1 cultured in the presence of the serum. Int. J. Immunopharmacol. 1997, 19, 255–262. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [Green Version]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, W.; Payyappat, S.; Cassidy, M.; Besley, C.; Power, K. Novel crAssphage marker genes ascertain sewage pollution in a recreational lake receiving urban stormwater runoff. Water Res. 2018, 145, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe 2018, 24, 653–664.e6. [Google Scholar] [CrossRef] [Green Version]
- Stachler, E.; Akyon, B.; de Carvalho, N.A.; Ference, C.; Bibby, K. Correlation of crAssphage qPCR Markers with Culturable and Molecular Indicators of Human Fecal Pollution in an Impacted Urban Watershed. Environ. Sci. Technol. 2018, 52, 7505–7512. [Google Scholar] [CrossRef]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J.; et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.P.; Chopera, D.; Havyarimana, E.; Wendoh, J.; Jaumdally, S.; Nyangahu, D.D.; Gray, C.M.; Martin, D.P.; Varsani, A.; Jaspan, H.B. crAssphage genomes identified in fecal samples of an adult and infants with evidence of positive genomic selective pressure within tail protein genes. Virus Res. 2021, 292, 198219. [Google Scholar] [CrossRef]
- Malla, B.; Ghaju Shrestha, R.; Tandukar, S.; Sherchand, J.B.; Haramoto, E. Performance Evaluation of Human-Specific Viral Markers and Application of Pepper Mild Mottle Virus and CrAssphage to Environmental Water Samples as Fecal Pollution Markers in the Kathmandu Valley, Nepal. Food Environ. Virol. 2019, 11, 274–287. [Google Scholar] [CrossRef]
- Li, Y.; Gordon, E.; Shean, R.C.; Idle, A.; Deng, X.; Greninger, A.L.; Delwart, E. CrAssphage and its bacterial host in cat feces. Sci. Rep. 2021, 11, 815. [Google Scholar] [CrossRef]
- García-Aljaro, C.; Ballesté, E.; Muniesa, M.; Jofre, J. Determination of crAssphage in water samples and applicability for tracking human faecal pollution. Microb. Biotechnol. 2017, 10, 1775–1780. [Google Scholar] [CrossRef]
- Stachler, E.; Kelty, C.; Sivaganesan, M.; Li, X.; Bibby, K.; Shanks, O.C. Quantitative CrAssphage PCR Assays for Human Fecal Pollution Measurement. Environ. Sci. Technol. 2017, 51, 9146–9154. [Google Scholar] [CrossRef]
- Park, G.W.; Ng, T.F.F.; Freeland, A.L.; Marconi, V.C.; Boom, J.A.; Staat, M.A.; Montmayeur, A.M.; Browne, H.; Narayanan, J.; Payne, D.C.; et al. CrAssphage as a Novel Tool to Detect Human Fecal Contamination on Environmental Surfaces and Hands. Emerg. Infect. Dis. 2020, 26, 1731–1739. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample Type a | No. of Raw Reads | % of Viral/Non-Viral Reads | Best BLASTN Hit b | Non-Assembled Reads c | Assembled Reads | Contig Length Range (nt) | Contig Identity d | Genome Coverage e |
|---|---|---|---|---|---|---|---|---|
| FBS1 MEX | 772,754 | 0.004/99.996 | Bovine parvovirus 3 | 18 | 4 | 181–311 | 93.7% | 28.2% |
| Bovine polyomavirus 1 | 2 | 1 | 205 | 99.0% | 4.7% | |||
| Moloney murine leukemia virus | 12 | 1 | 206 | 99.5% | 3.3% | |||
| FBS2 USA | 1,538,378 | 0.117/99.882 | Bovine parvovirus 2 | 1138 | 43 | 107–1904 | 97.7% | 100% |
| Bovine parvovirus 3 | 3 | 1 | 219 | 89.0% | 8.6% | |||
| Bovine polyomavirus 1 | 660 | 21 | 106–2250 | 100% | 99.4% | |||
| Bovine viral diarrhea virus 1 | 5 | 2 | 129–180 | 93.0% | 2.5% | |||
| Bovine viral diarrhea virus 2 | 6 | 2 | 151–224 | 94.7% | 2.4% | |||
| NCS1 USA | 3,157,462 | 0.453/99.546 | Bovine bocaparvovirus 2 | 878 | 16 | 122–3110 | 100% | 88.2% |
| Bosavirus | 1806 | 34 | 101–3021 | 100% | 100% | |||
| Bovine hokovirus 1 | 41 | 4 | 120–2179 | 5.0% | 46.9% | |||
| Bovine hokovirus 2 | 39 | 1 | 395 | 98.1% | 45.6% | |||
| Bovine kobuvirus | 4 | 2 | 151–207 | 95.4% | 4.4% | |||
| Bovine parvovirus 1 | 8 | 1 | 420 | 96.0% | 12.8% | |||
| Bovine parvovirus 2 | 11,470 | 297 | 101–2552 | 100% | 100% | |||
| Bovine parvovirus 3 | 67 | 13 | 113–623 | 97.7% | 59.2% | |||
| Cyclovirus Equ1 | 4 | 2 | 103–209 | 100% | 17.5% | |||
| Feces associated gemycircularvirus 14 | 2 | 1 | 192 | 97.4% | 9.4% | |||
| Gopherus associated genomovirus 1 | 2 | 1 | 181 | 82.3% | 10.1% | |||
| Mongoose feces-associated gemycircularvirus b | 2 | 1 | 181 | 86.2% | 8.2% | |||
| Po-Circo-like virus 21 | 2 | 1 | 246 | 93.3% | 6.2% | |||
| Porcine associated porprismacovirus 10 | 2 | 1 | 151 | 88.7% | 5.7% | |||
| NCS2 NZ | 2,806,994 | 35.240/64.759 | Bosavirus | 18 | 4 | 157–411 | 100% | 26.7% |
| Bovine parvovirus 2 | 44 | 11 | 281–1951 | 86.0% | 25.3% | |||
| Bovine parvovirus 3 | 989,123 | 7700 | 100–2471 | 100% | 100% | |||
| Bovine rotavirus A | 5 | 2 | 113–157 | 97.3% | 7.7% | |||
| Starling circovirus | 2 | 1 | 188 | 95.2% | 9.1% | |||
| NCS3 NZ | 1,082,206 | 2.081/97.918 | Bovine bocaparvovirus 2 | 4 | 1 | 245 | 100% | 5.0% |
| Bosavirus | 20,458 | 365 | 100–2050 | 100% | 100% | |||
| Bovine adenovirus 4 | 4 | 2 | 103–147 | 99.3% | 0.8% | |||
| Bovine parvovirus 2 | 2002 | 232 | 101–1370 | 96.6% | 100% | |||
| Bovine polyomavirus 1 | 10 | 2 | 141–221 | 100% | 8.6% | |||
| Bovine rotavirus A | 4 | 1 | 298 | 95.3% | 9.0% | |||
| Feces associated gemycircularvirus 14 | 10 | 3 | 190–238 | 99.6% | 37.7% | |||
| Feces associated gemycircularvirus 16 | 2 | 1 | 140 | 92.1% | 6.3% | |||
| Gopherus associated genomovirus 1 | 2 | 1 | 200 | 79.0% | 9.2% | |||
| Human rotavirus A | 4 | 1 | 103 | 96.1% | 4.0% | |||
| Mongoose feces-associated gemycircularvirus a | 2 | 1 | 176 | 80.7% | 8.4% | |||
| Mongoose feces-associated gemycircularvirus b | 22 | 4 | 387–571 | 99.2% | 88.2% | |||
| Sewage-associated gemycircularvirus 3 | 2 | 1 | 162 | 98.8% | 7.6% | |||
| NCS4 USA | 1,099,344 | 0.021/99.978 | Bovine hokovirus 1 | 2 | 1 | 233 | 88.6% | 4.6% |
| Bovine kobuvirus | 2 | 1 | 203 | 94.1% | 2.5% | |||
| Bovine parvovirus 2 | 217 | 26 | 149–1679 | 98.4% | 100% | |||
| Bovine polyomavirus | 6 | 2 | 115–233 | 97.6% | 13.0% | |||
| Bovine serum-associated circular virus | 4 | 1 | 370 | 92.0% | 73.0% | |||
| Genomoviridae sp. ctca367 | 2 | 1 | 119 | 96.6% | 5.4% | |||
| BCS1 USA | 1,035,018 | 41.878/58.121 | Bovine adenovirus 6 | 6 | 2 | 280–282 | 98.9% | 2.9% |
| Bovine hokovirus 1 | 34 | 7 | 107–184 | 100% | 15.3% | |||
| Bovine hokovirus 2 | 15,009 | 93 | 105–1418 | 100% | 100% | |||
| Bovine parvovirus 2 | 149,979 | 3733 | 100–2888 | 100% | 100% | |||
| Bovine parvovirus 3 | 268,265 | 3569 | 100–2574 | 100% | 100% | |||
| Bo-Circo-like virus CH | 6 | 1 | 149 | 100% | 3.8% | |||
| Circovirus sp. PoCirV VIRES YN02 C2 | 3 | 1 | 334 | 91.3% | 31.1% | |||
| Po-Circo-like virus 21 | 12 | 3 | 132–252 | 95.5% | 14.4% | |||
| Po-Circo-like virus 22 | 13 | 1 | 192 | 88.5% | 4.9% | |||
| Po-Circo-like virus 41 | 3 | 1 | 191 | 93.6% | 6.5% | |||
| Po-Circo-like virus S20 | 7 | 1 | 262 | 96.5% | 38.9% | |||
| Po-Circo-like virus GX19 | 11 | 1 | 480 | 90.2% | 12.2% |
| Sample Type a | Number of Contigs | Contig Length Range | Best BLASTN Hit ID of the Longest Contig b/Sample Type/Species | Contig Identity c |
|---|---|---|---|---|
| FBS1 Mex | 0 | Not apply | Not apply | Not apply |
| FBS2 USA | 1 | 80 | Uncultured crAssphage -NC_024711/Fecal/human | 86.08% |
| NCS1 USA | 69 | 76-625 | CrAssphage FA1-2_000172F-MK415404/Gut/human | 99.20% |
| NCS2 NZ | 56 | 108–409 | CrAssphage YS1-2_2437-MK415410/Gut/human | 98.78% |
| NCS3 NZ | 107 | 64–721 | CrAssphage LMMB-MT006214/Fecal/human | 94.59% |
| NCS4 USA | 8 | 100–241 | CrAssphage sp. C0531BW4-MW067003/Fecal/human | 97.93% |
| BCS1 USA | 1 | 74 | CrAssphage YS1-2_2437-MK415410/Gut/human | 82.43% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paim, W.P.; Maggioli, M.F.; Falkenberg, S.M.; Ramachandran, A.; Weber, M.N.; Canal, C.W.; Bauermann, F.V. Virome Characterization in Commercial Bovine Serum Batches—A Potentially Needed Testing Strategy for Biological Products. Viruses 2021, 13, 2425. https://doi.org/10.3390/v13122425
Paim WP, Maggioli MF, Falkenberg SM, Ramachandran A, Weber MN, Canal CW, Bauermann FV. Virome Characterization in Commercial Bovine Serum Batches—A Potentially Needed Testing Strategy for Biological Products. Viruses. 2021; 13(12):2425. https://doi.org/10.3390/v13122425
Chicago/Turabian StylePaim, Willian P., Mayara F. Maggioli, Shollie M. Falkenberg, Akhilesh Ramachandran, Matheus N. Weber, Cláudio W. Canal, and Fernando V. Bauermann. 2021. "Virome Characterization in Commercial Bovine Serum Batches—A Potentially Needed Testing Strategy for Biological Products" Viruses 13, no. 12: 2425. https://doi.org/10.3390/v13122425
APA StylePaim, W. P., Maggioli, M. F., Falkenberg, S. M., Ramachandran, A., Weber, M. N., Canal, C. W., & Bauermann, F. V. (2021). Virome Characterization in Commercial Bovine Serum Batches—A Potentially Needed Testing Strategy for Biological Products. Viruses, 13(12), 2425. https://doi.org/10.3390/v13122425