
ATM-Dependent Phosphorylation of Hepatitis B Core Protein in Response to Genotoxic Stress

 and
and 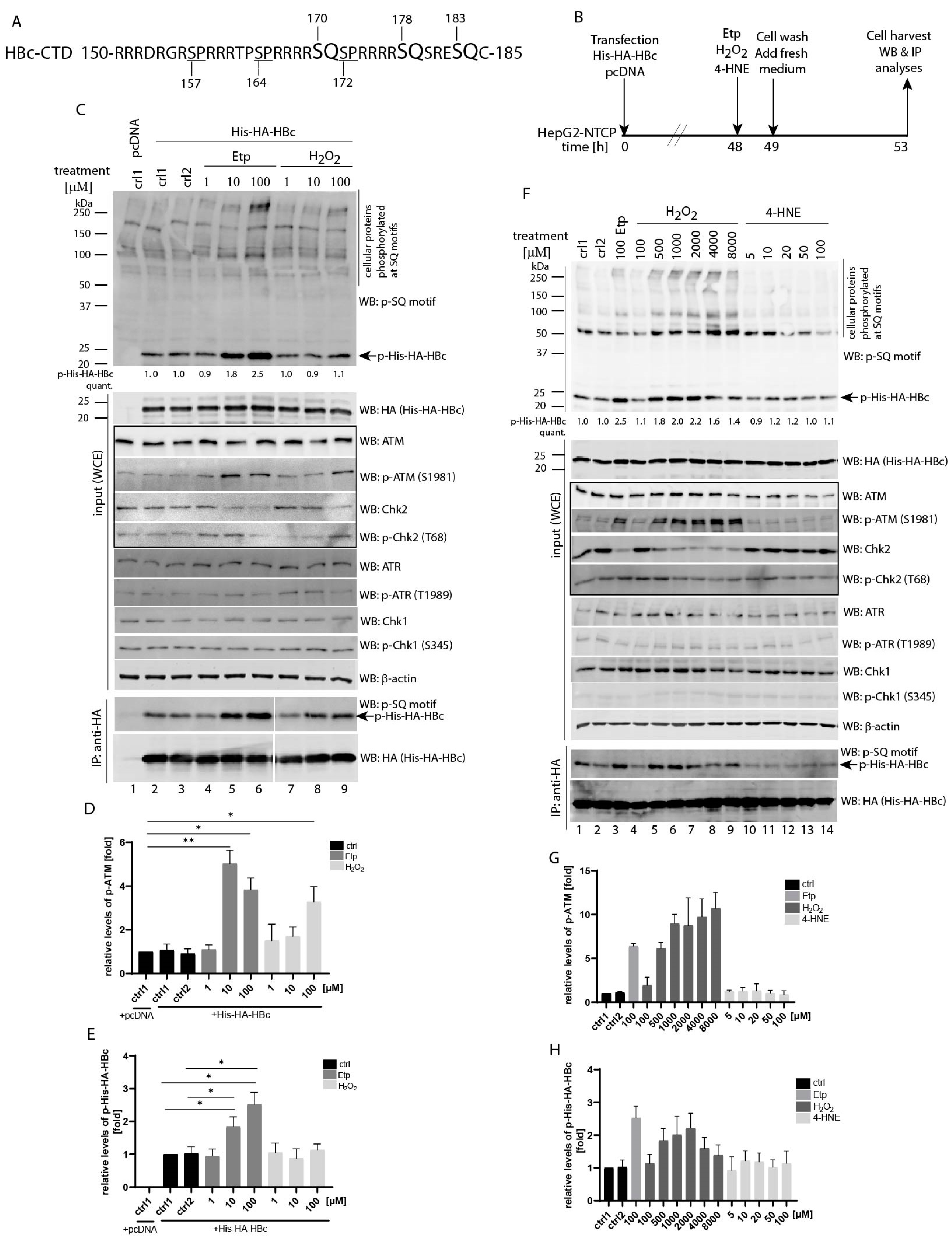{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Plasmids
2.3. Antibodies
2.4. SiRNAs
2.5. Chemicals and Inhibitors
2.6. HBV Infection of HepG2-NTCP Cells
2.7. Transfections
2.8. Preparation of Protein Samples
2.9. Quantification of HBc, ATM, Phospho-HBc (p-HBc) and Phospho-ATM (p-ATM)
2.10. HBeAg Detection by ELISA
2.11. Statistical Analysis
3. Results
3.1. Etoposide- and H2O2-Induced HBc Phosphorylation
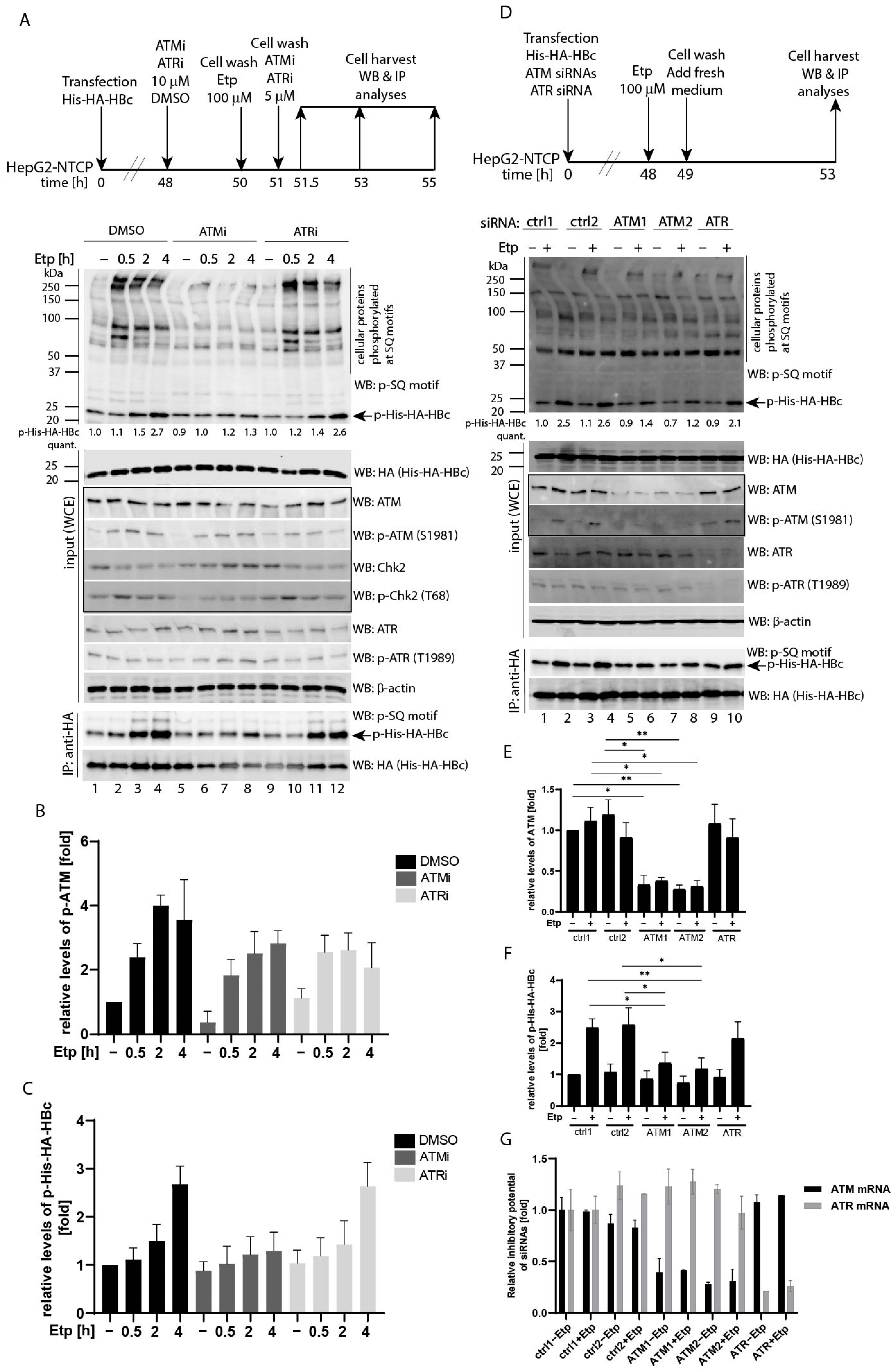
3.2. Etoposide-Induced Phosphorylation of HBc Is ATM-Dependent
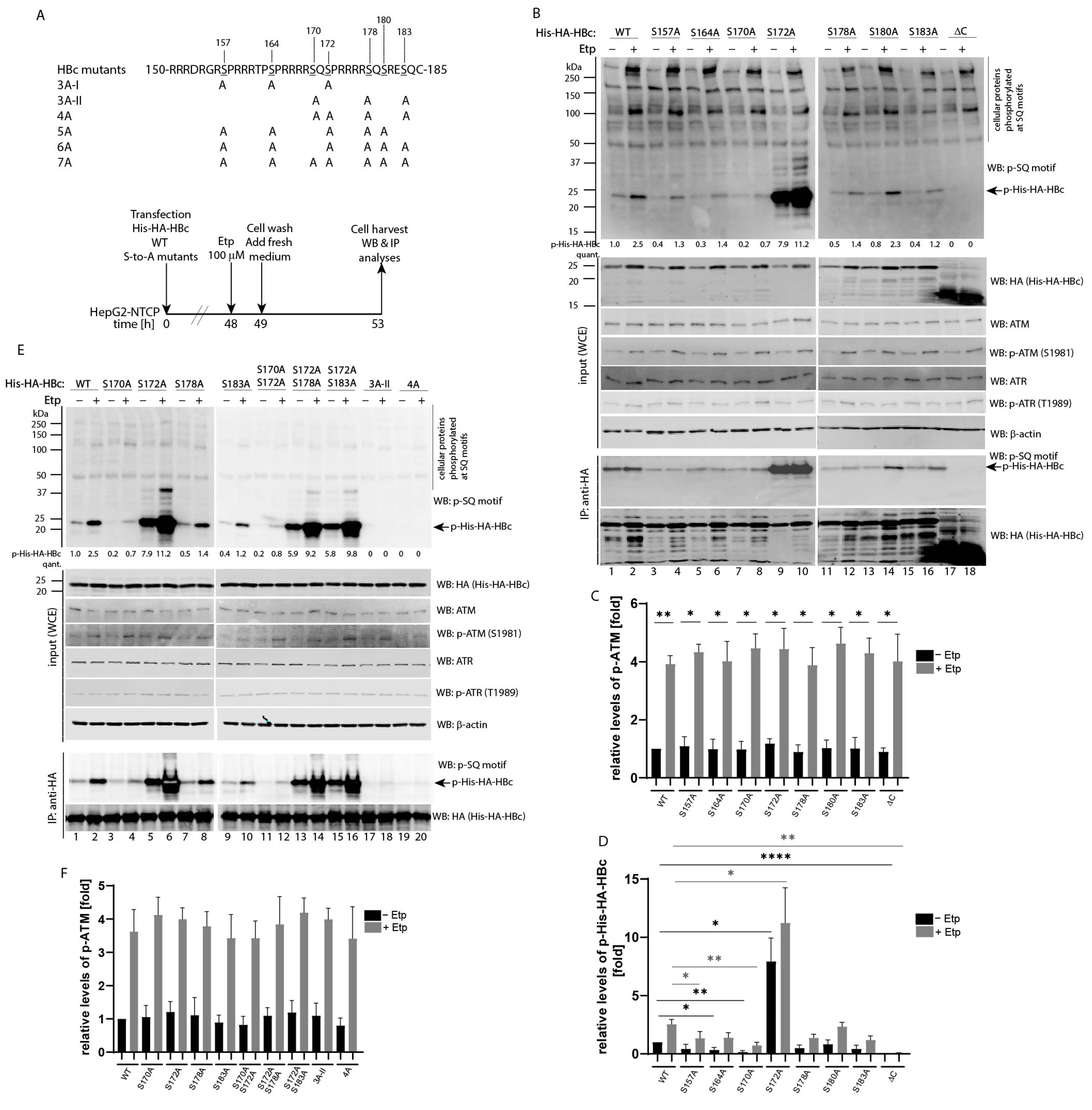
3.3. Mapping of Etoposide-Induced Phosphorylation Sites on HBc
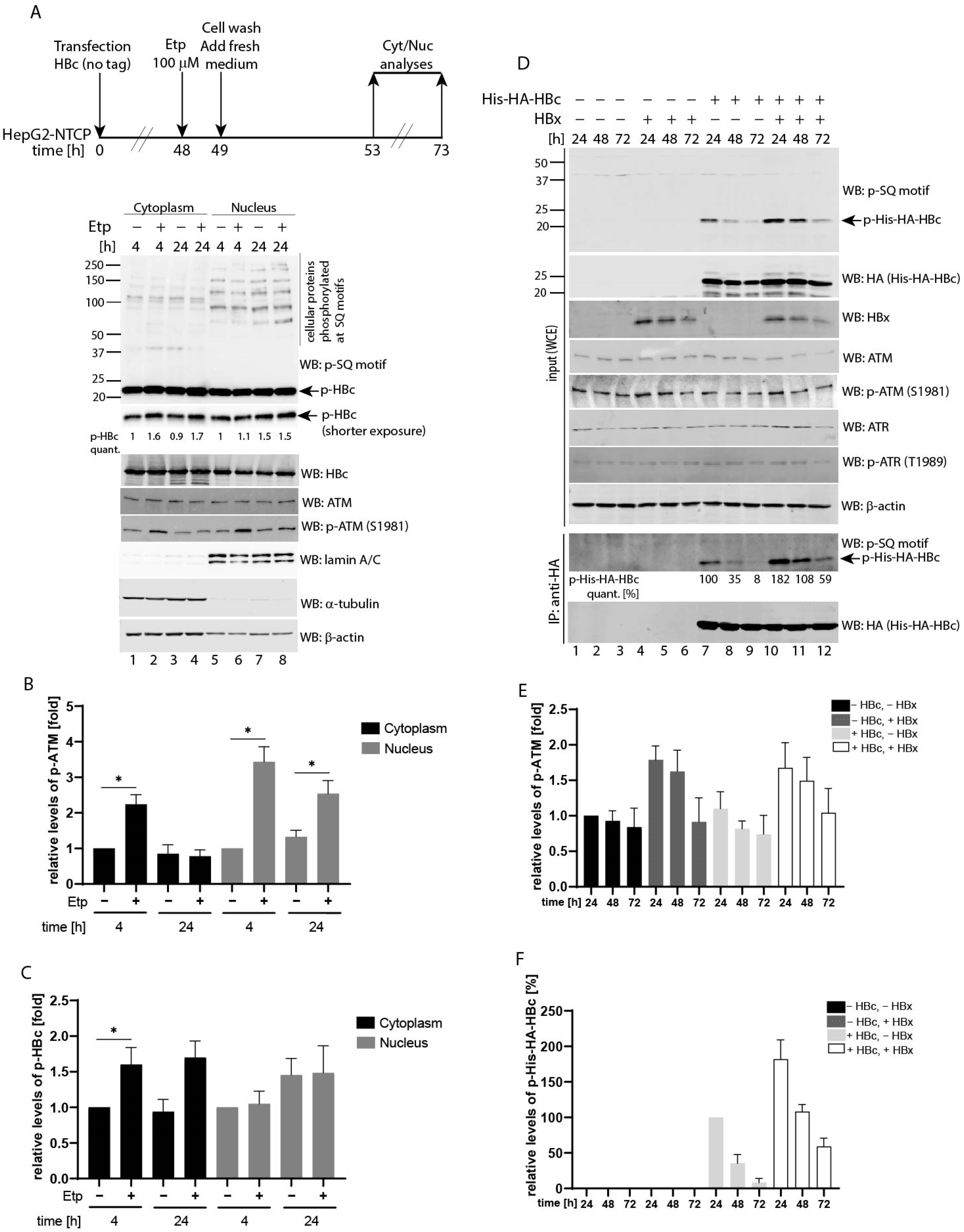
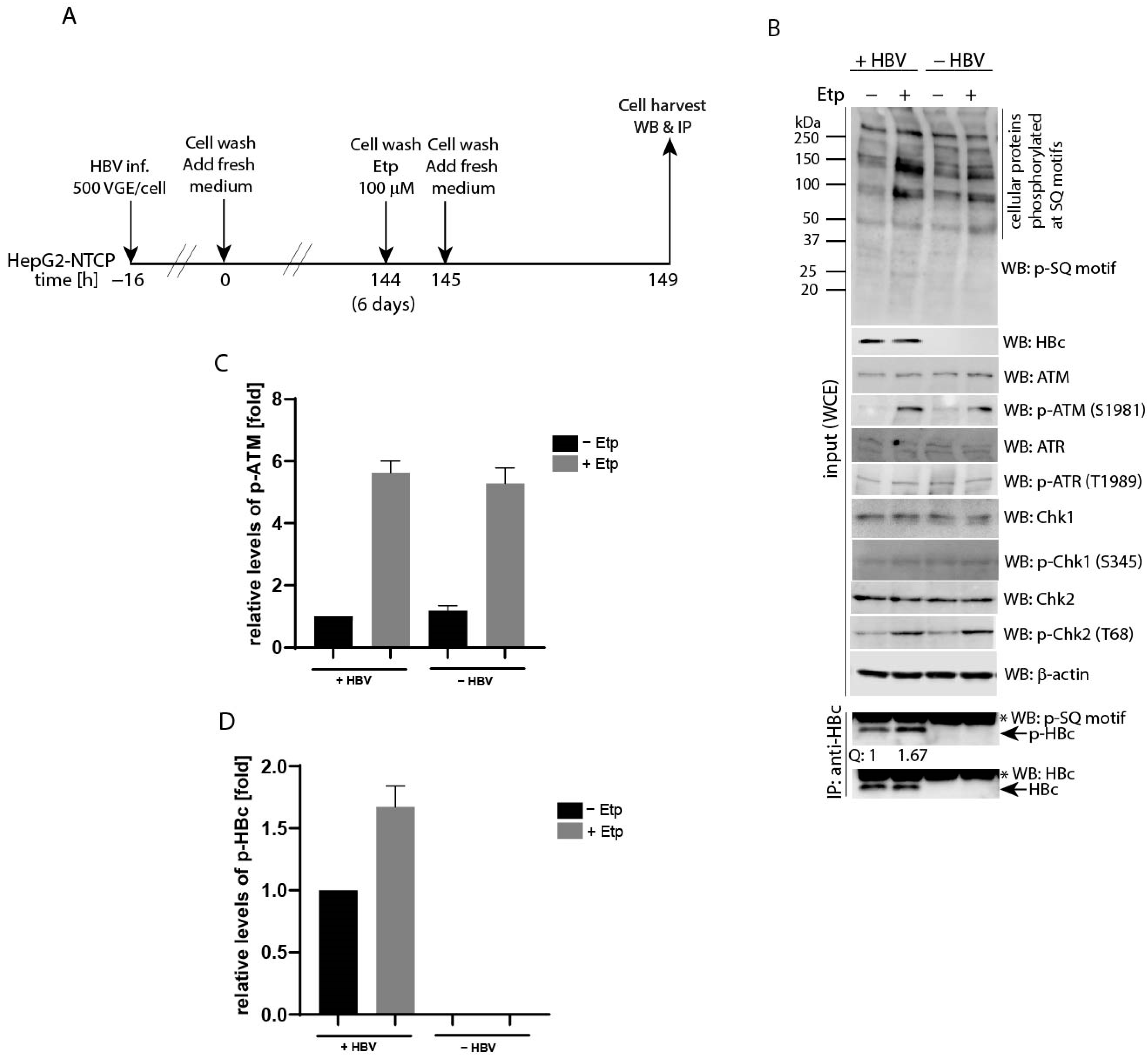
3.4. Etoposide-Induced Phosphorylation Occurs on Cytoplasmic HBc
3.5. Co-Expression of HBV X (HBx) Leads to Upregulated HBc Phosphorylation at SQ Motifs
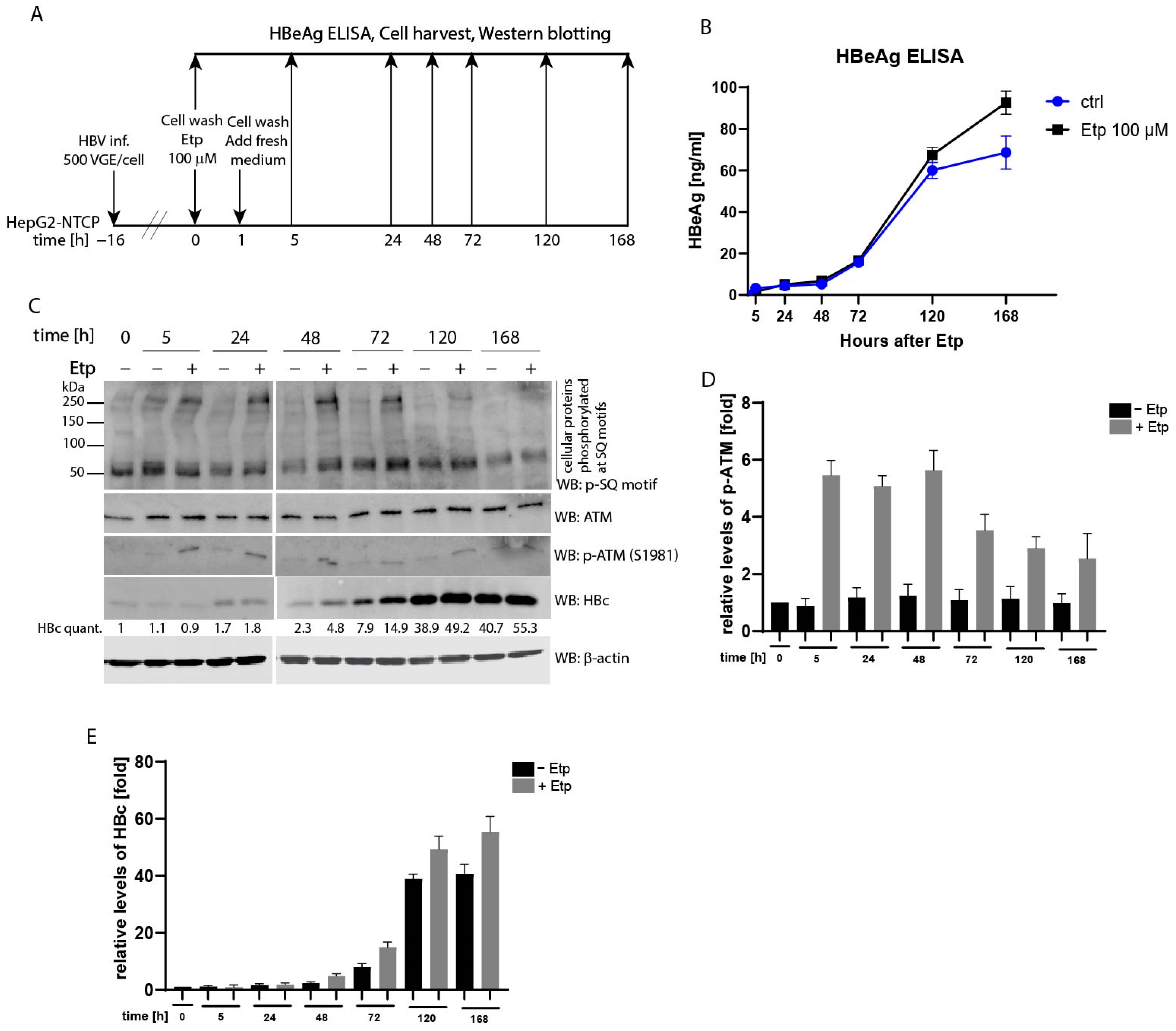
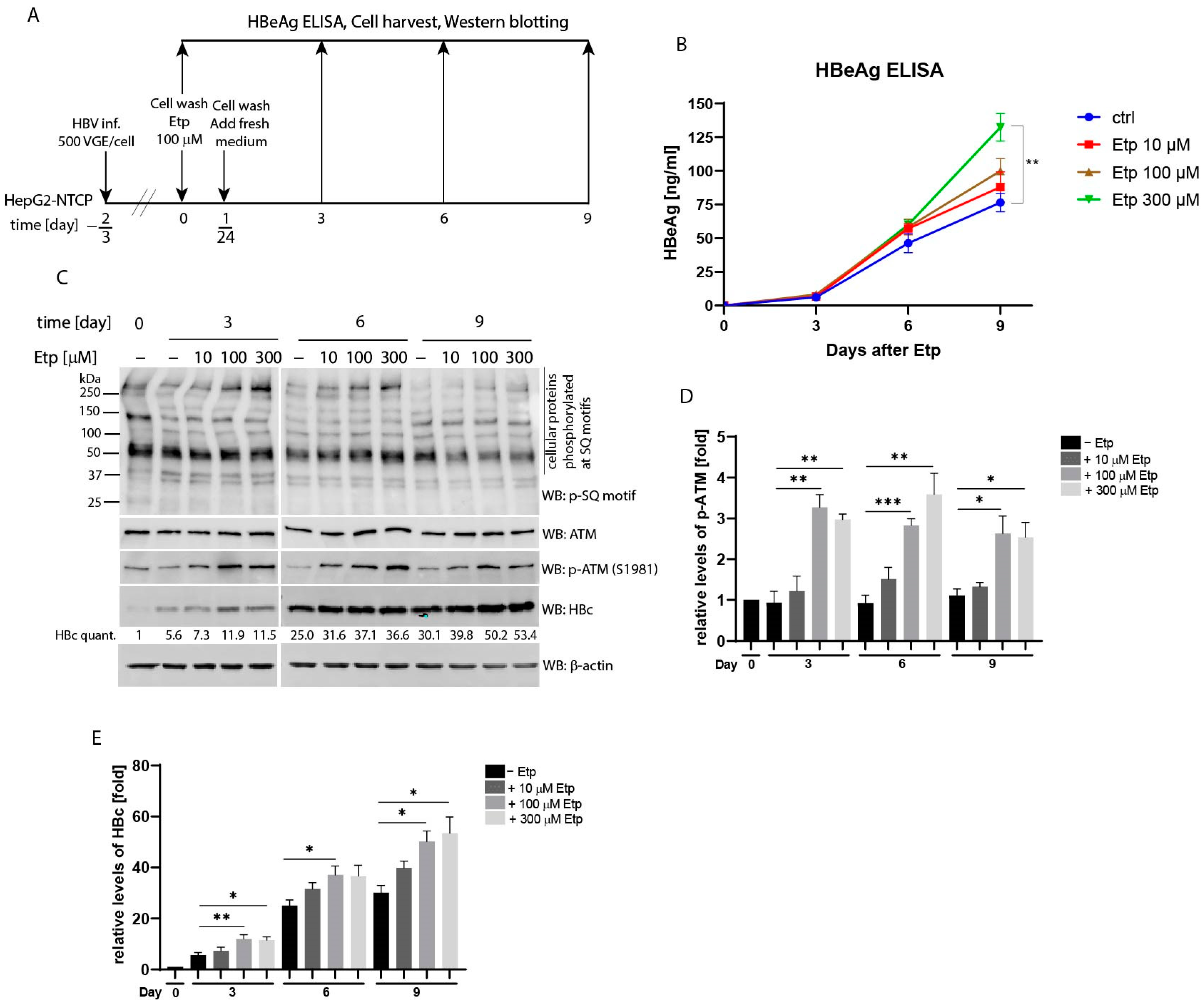
3.6. Effect of Etoposide Treatment on HBV Infection
4. Discussion
4.1. ATM-Dependent Phosphorylation of HBc at S170
4.2. The Role of “Major” Phosphorylation Sites in HBc Phosphorylation at SQ Motifs
4.3. Phosphorylation of HBc CTD and Its Role in HBV Life Cycle
4.4. The Role of Cytoplasmic ATM Pathway in the Regulation of HBc Phosphorylation
4.5. Interaction between HBV and Host ATM-Regulated Pathway
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 2004, 11, 97–107. [Google Scholar] [CrossRef]
- Torres, H.A.; Davila, M. Reactivation of hepatitis B virus and hepatitis C virus in patients with cancer. Nat. Rev. Clin. Oncol. 2012, 9, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Robinson, W.S. The genome of hepatitis B virus. Annu. Rev. Microbiol. 1977, 31, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diab, A.; Foca, A.; Zoulim, F.; Durantel, D.; Andrisani, O. The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: Implications for the development of HBc-targeting antivirals. Antivir. Res. 2018, 149, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.K.; Cheng, C.Y.S.; Tsoi, S.Y.J.; Huang, F.Y.; Liu, F.; Seto, W.K.; Lai, C.L.; Yuen, M.F.; Wong, D.K. Role of hepatitis B core protein in HBV transcription and recruitment of histone acetyltransferases to cccDNA minichromosome. Antivir. Res. 2017, 144, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zlotnick, A.; Venkatakrishnan, B.; Tan, Z.; Lewellyn, E.; Turner, W.; Francis, S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antivir. Res. 2015, 121, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnbaum, F.; Nassal, M. Hepatitis B virus nucleocapsid assembly: Primary structure requirements in the core protein. J. Virol. 1990, 64, 3319–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallina, A.; Bonelli, F.; Zentilin, L.; Rindi, G.; Muttini, M.; Milanesi, G. A recombinant hepatitis B core antigen polypeptide with the protamine-like domain deleted self-assembles into capsid particles but fails to bind nucleic acids. J. Virol. 1989, 63, 4645–4652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, T.H.; Liou, A.T.; Su, P.Y.; Wu, H.N.; Shih, C. Nucleic acid chaperone activity associated with the arginine-rich domain of human hepatitis B virus core protein. J. Virol. 2014, 88, 2530–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.C.; Huang, E.Y.; Su, P.Y.; Wu, S.Y.; Yang, C.C.; Lin, Y.S.; Chang, W.C.; Shih, C. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog. 2010, 6, e1001162. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.C.; Li, H.C.; Shih, C. A Homokaryon Assay for Nucleocytoplasmic Shuttling Activity of HBV Core Protein. Methods Mol. Biol. 2017, 1540, 53–58. [Google Scholar] [CrossRef] [PubMed]
- de Rocquigny, H.; Rat, V.; Pastor, F.; Darlix, J.L.; Hourioux, C.; Roingeard, P. Phosphorylation of the Arginine-Rich C-Terminal Domains of the Hepatitis B Virus (HBV) Core Protein as a Fine Regulator of the Interaction between HBc and Nucleic Acid. Viruses 2020, 12, 738. [Google Scholar] [CrossRef]
- Su, P.Y.; Yang, C.J.; Chu, T.H.; Chang, C.H.; Chiang, C.; Tang, F.M.; Lee, C.Y.; Shih, C. HBV maintains electrostatic homeostasis by modulating negative charges from phosphoserine and encapsidated nucleic acids. Sci. Rep. 2016, 6, 38959. [Google Scholar] [CrossRef] [Green Version]
- Daub, H.; Blencke, S.; Habenberger, P.; Kurtenbach, A.; Dennenmoser, J.; Wissing, J.; Ullrich, A.; Cotten, M. Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J. Virol. 2002, 76, 8124–8137. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Hwang, S.G.; Chwae, Y.J.; Park, S.; Shin, H.J.; Kim, K. Phosphoacceptors threonine 162 and serines 170 and 178 within the carboxyl-terminal RRRS/T motif of the hepatitis B virus core protein make multiple contributions to hepatitis B virus replication. J. Virol. 2014, 88, 8754–8767. [Google Scholar] [CrossRef] [Green Version]
- Melegari, M.; Wolf, S.K.; Schneider, R.J. Hepatitis B virus DNA replication is coordinated by core protein serine phosphorylation and HBx expression. J. Virol. 2005, 79, 9810–9820. [Google Scholar] [CrossRef] [Green Version]
- Heger-Stevic, J.; Zimmermann, P.; Lecoq, L.; Bottcher, B.; Nassal, M. Hepatitis B virus core protein phosphorylation: Identification of the SRPK1 target sites and impact of their occupancy on RNA binding and capsid structure. PLoS Pathog. 2018, 14, e1007488. [Google Scholar] [CrossRef] [Green Version]
- Ludgate, L.; Liu, K.; Luckenbaugh, L.; Streck, N.; Eng, S.; Voitenleitner, C.; Delaney, W.E.T.; Hu, J. Cell-Free Hepatitis B Virus Capsid Assembly Dependent on the Core Protein C-Terminal Domain and Regulated by Phosphorylation. J. Virol. 2016, 90, 5830–5844. [Google Scholar] [CrossRef] [Green Version]
- Yang, F. Post-translational Modification Control of HBV Biological Processes. Front. Microbiol. 2018, 9, 2661. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Foca, A.; Fusil, F.; Lahlali, T.; Jalaguier, P.; Amirache, F.; N’Guyen, L.; Isorce, N.; Cosset, F.L.; Zoulim, F.; et al. Polo-like-kinase 1 is a proviral host factor for hepatitis B virus replication. Hepatology 2017, 66, 1750–1765. [Google Scholar] [CrossRef]
- Langerova, H.; Lubyova, B.; Zabransky, A.; Hubalek, M.; Glendova, K.; Aillot, L.; Hodek, J.; Strunin, D.; Janovec, V.; Hirsch, I.; et al. Hepatitis B Core Protein Is Post-Translationally Modified through K29-Linked Ubiquitination. Cells 2020, 9, 2547. [Google Scholar] [CrossRef] [PubMed]
- Lubyova, B.; Hodek, J.; Zabransky, A.; Prouzova, H.; Hubalek, M.; Hirsch, I.; Weber, J. PRMT5: A novel regulator of Hepatitis B virus replication and an arginine methylase of HBV core. PLoS ONE 2017, 12, e0186982. [Google Scholar] [CrossRef]
- Lubyova, B.; Weber, J. Posttranslational modifications of HBV core protein. Acta Virol. 2020, 64, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, R.T. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair 2004, 3, 883–887. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair 2009, 8, 1004–1008. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Hou, N.B.; Song, T.; He, X.; Zheng, Z.R.; Ma, Q.J.; Li, L.; Zhang, Y.H.; Zhong, H. Cellular DNA repair cofactors affecting hepatitis B virus infection and replication. World J. Gastroenterol. 2008, 14, 5059–5065. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Hou, N.B.; Yang, X.L.; He, X.; Liu, Y.; Zhang, Y.H.; Wei, C.W.; Song, T.; Li, L.; Ma, Q.J.; et al. Ataxia telangiectasia-mutated-Rad3-related DNA damage checkpoint signaling pathway triggered by hepatitis B virus infection. World J. Gastroenterol. 2008, 14, 6163–6170. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.S.; Ji, J.H.; Cho, M.Y.; Yoo, Y.S.; Park, Y.Y.; Cha, H.J.; Lee, Y.; Kim, Y.; Cho, H. Hepatitis B virus X protein activates the ATM-Chk2 pathway and delays cell cycle progression. J. Gen. Virol. 2015, 96, 2242–2251. [Google Scholar] [CrossRef] [PubMed]
- Kostyusheva, A.; Brezgin, S.; Bayurova, E.; Gordeychuk, I.; Isaguliants, M.; Goptar, I.; Urusov, F.; Nikiforova, A.; Volchkova, E.; Kostyushev, D.; et al. ATM and ATR Expression Potentiates HBV Replication and Contributes to Reactivation of HBV Infection upon DNA Damage. Viruses 2019, 11, 997. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Luckenbaugh, L.; Hu, H.; Yan, Z.; Gao, L.; Hu, J. Involvement of Host ATR-CHK1 Pathway in Hepatitis B Virus Covalently Closed Circular DNA Formation. MBio 2020, 11, e03423-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartek, J.; Lukas, J. DNA repair: Damage alert. Nature 2003, 421, 486–488. [Google Scholar] [CrossRef]
- Chaudhary, P.; Sharma, R.; Sahu, M.; Vishwanatha, J.K.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal induces G2/M phase cell cycle arrest by activation of the ataxia telangiectasia mutated and Rad3-related protein (ATR)/checkpoint kinase 1 (Chk1) signaling pathway. J. Biol. Chem. 2013, 288, 20532–20546. [Google Scholar] [CrossRef] [Green Version]
- Everett, R.D. Interactions between DNA viruses, ND10 and the DNA damage response. Cell Microbiol. 2006, 8, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Weitzman, M.D. Keeping viruses in Chk: DNA damage signaling puts the brakes on transformation. Cell Host Microbe 2010, 8, 464–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA Replication and the Host DNA Damage Response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, N.J.; Price, A.M.; Weitzman, M.D. Take your PIKK: Tumour viruses and DNA damage response pathways. Philos. Trans. R Soc. Lond B Biol. Sci. 2017, 372, 20160269. [Google Scholar] [CrossRef] [Green Version]
- Xi, J.; Luckenbaugh, L.; Hu, J. Multiple roles of PP2A binding motif in hepatitis B virus core linker and PP2A in regulating core phosphorylation state and viral replication. PLoS Pathog. 2021, 17, e1009230. [Google Scholar] [CrossRef]
- Basagoudanavar, S.H.; Perlman, D.H.; Hu, J. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J. Virol. 2007, 81, 1641–1649. [Google Scholar] [CrossRef] [Green Version]
- Gazina, E.V.; Fielding, J.E.; Lin, B.; Anderson, D.A. Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J. Virol. 2000, 74, 4721–4728. [Google Scholar] [CrossRef] [Green Version]
- Lewellyn, E.B.; Loeb, D.D. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J. Virol. 2011, 85, 1298–1309. [Google Scholar] [CrossRef] [Green Version]
- Kock, J.; Nassal, M.; Deres, K.; Blum, H.E.; von Weizsacker, F. Hepatitis B virus nucleocapsids formed by carboxy-terminally mutated core proteins contain spliced viral genomes but lack full-size DNA. J. Virol. 2004, 78, 13812–13818. [Google Scholar] [CrossRef] [Green Version]
- Le Pogam, S.; Chua, P.K.; Newman, M.; Shih, C. Exposure of RNA templates and encapsidation of spliced viral RNA are influenced by the arginine-rich domain of human hepatitis B virus core antigen (HBcAg 165–173). J. Virol. 2005, 79, 1871–1887. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.; Chua, P.K.; Tang, F.M.; Su, P.Y.; Shih, C. Testing an electrostatic interaction hypothesis of hepatitis B virus capsid stability by using an in vitro capsid disassembly/reassembly system. J. Virol. 2009, 83, 10616–10626. [Google Scholar] [CrossRef] [Green Version]
- Chua, P.K.; Tang, F.M.; Huang, J.Y.; Suen, C.S.; Shih, C. Testing the balanced electrostatic interaction hypothesis of hepatitis B virus DNA synthesis by using an in vivo charge rebalance approach. J. Virol. 2010, 84, 2340–2351. [Google Scholar] [CrossRef] [Green Version]
- Hatton, T.; Zhou, S.; Standring, D.N. RNA- and DNA-binding activities in hepatitis B virus capsid protein: A model for their roles in viral replication. J. Virol. 1992, 66, 5232–5241. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Luckenbaugh, L.; Bruss, V.; Hu, J. Alteration of Mature Nucleocapsid and Enhancement of Covalently Closed Circular DNA Formation by Hepatitis B Virus Core Mutants Defective in Complete-Virion Formation. J. Virol. 2015, 89, 10064–10072. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Ban, H.; Zheng, H.; Liu, M.; Chang, J.; Guo, J.T. Protein phosphatase 1 catalyzes HBV core protein dephosphorylation and is co-packaged with viral pregenomic RNA into nucleocapsids. PLoS Pathog. 2020, 16, e1008669. [Google Scholar] [CrossRef]
- Ludgate, L.; Ning, X.; Nguyen, D.H.; Adams, C.; Mentzer, L.; Hu, J. Cyclin-dependent kinase 2 phosphorylates s/t-p sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J. Virol. 2012, 86, 12237–12250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Xi, J.; Gao, L.; Hu, J. Role of Hepatitis B virus capsid phosphorylation in nucleocapsid disassembly and covalently closed circular DNA formation. PLoS Pathog. 2020, 16, e1008459. [Google Scholar] [CrossRef] [Green Version]
- Okabe, M.; Enomoto, M.; Maeda, H.; Kuroki, K.; Ohtsuki, K. Biochemical characterization of suramin as a selective inhibitor for the PKA-mediated phosphorylation of HBV core protein in vitro. Biol. Pharm. Bull. 2006, 29, 1810–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittkop, L.; Schwarz, A.; Cassany, A.; Grun-Bernhard, S.; Delaleau, M.; Rabe, B.; Cazenave, C.; Gerlich, W.; Glebe, D.; Kann, M. Inhibition of protein kinase C phosphorylation of hepatitis B virus capsids inhibits virion formation and causes intracellular capsid accumulation. Cell Microbiol. 2010, 12, 962–975. [Google Scholar] [CrossRef]
- Barlow, C.; Ribaut-Barassin, C.; Zwingman, T.A.; Pope, A.J.; Brown, K.D.; Owens, J.W.; Larson, D.; Harrington, E.A.; Haeberle, A.M.; Mariani, J.; et al. ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc. Natl. Acad. Sci. USA 2000, 97, 871–876. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.S.; Kirsch, D.G.; Canman, C.E.; Ahn, J.H.; Ziv, Y.; Newman, L.S.; Darnell, R.B.; Shiloh, Y.; Kastan, M.B. ATM binds to beta-adaptin in cytoplasmic vesicles. Proc. Natl. Acad. Sci. USA 1998, 95, 10146–10151. [Google Scholar] [CrossRef] [Green Version]
- Oka, A.; Takashima, S. Expression of the ataxia-telangiectasia gene (ATM) product in human cerebellar neurons during development. Neurosci. Lett. 1998, 252, 195–198. [Google Scholar] [CrossRef]
- Watters, D.; Kedar, P.; Spring, K.; Bjorkman, J.; Chen, P.; Gatei, M.; Birrell, G.; Garrone, B.; Srinivasa, P.; Crane, D.I.; et al. Localization of a portion of extranuclear ATM to peroxisomes. J. Biol. Chem. 1999, 274, 34277–34282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Stagni, V.; Ferri, A.; Cirotti, C.; Barila, D. ATM Kinase-Dependent Regulation of Autophagy: A Key Player in Senescence? Front Cell Dev. Biol. 2020, 8, 599048. [Google Scholar] [CrossRef]
- Fu, X.; Wan, S.; Lyu, Y.L.; Liu, L.F.; Qi, H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS ONE 2008, 3, e2009. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Halicka, H.D.; Traganos, F.; Seiter, K.; Darzynkiewicz, Z. Induction of ATM activation, histone H2AX phosphorylation and apoptosis by etoposide: Relation to cell cycle phase. Cell Cycle 2007, 6, 371–376. [Google Scholar] [CrossRef]
- Zhang, A.; Lyu, Y.L.; Lin, C.P.; Zhou, N.; Azarova, A.M.; Wood, L.M.; Liu, L.F. A protease pathway for the repair of topoisomerase II-DNA covalent complexes. J. Biol. Chem. 2006, 281, 35997–36003. [Google Scholar] [CrossRef] [Green Version]
- Slagle, B.L.; Andrisani, O.M.; Bouchard, M.J.; Lee, C.G.; Ou, J.H.; Siddiqui, A. Technical standards for hepatitis B virus X protein (HBx) research. Hepatology 2015, 61, 1416–1424. [Google Scholar] [CrossRef] [Green Version]
- Aleem, A.; Al Amoudi, S.; Al-Mashhadani, S.; Siddiqui, N. Haemophagocytic syndrome associated with hepatitis-B virus infection responding to etoposide. Clin. Lab. Haematol. 2005, 27, 395–398. [Google Scholar] [CrossRef]
- Cheng, J.C.; Liu, M.C.; Tsai, S.Y.; Fang, W.T.; Jer-Min Jian, J.; Sung, J.L. Unexpectedly frequent hepatitis B reactivation by chemoradiation in postgastrectomy patients. Cancer 2004, 101, 2126–2133. [Google Scholar] [CrossRef]
- Zaman, S.; Melia, W.; Johnson, P.; White, Y.; Williams, R. Effect of cytotoxic chemotherapy on hepatitis B viral markers in patients with hepatocellular carcinoma. Clin. Oncol. 1984, 10, 247–252. [Google Scholar]
- Niitsu, N.; Hagiwara, Y.; Tanae, K.; Kohri, M.; Takahashi, N. Prospective analysis of hepatitis B virus reactivation in patients with diffuse large B-cell lymphoma after rituximab combination chemotherapy. J. Clin. Oncol. 2010, 28, 5097–5100. [Google Scholar] [CrossRef] [PubMed]
- Faggioli, P.; De Paschale, M.; Tocci, A.; Luoni, M.; Fava, S.; De Paoli, A.; Tosi, A.; Cassi, E. Acute hepatic toxicity during cyclic chemotherapy in non Hodgkin’s lymphoma. Haematologica 1997, 82, 38–42. [Google Scholar] [PubMed]
- Dansako, H.; Ueda, Y.; Satoh, S.; Kato, N. Extracellular vesicles activate ATM-Chk2 signaling pathway through the intercellular transfer of mitochondrial DNA in HBV-infected human hepatocytes. FASEB J. 2021, 35, e21680. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lubyova, B.; Tikalova, E.; Krulova, K.; Hodek, J.; Zabransky, A.; Hirsch, I.; Weber, J. ATM-Dependent Phosphorylation of Hepatitis B Core Protein in Response to Genotoxic Stress. Viruses 2021, 13, 2438. https://doi.org/10.3390/v13122438
Lubyova B, Tikalova E, Krulova K, Hodek J, Zabransky A, Hirsch I, Weber J. ATM-Dependent Phosphorylation of Hepatitis B Core Protein in Response to Genotoxic Stress. Viruses. 2021; 13(12):2438. https://doi.org/10.3390/v13122438
Chicago/Turabian StyleLubyova, Barbora, Eva Tikalova, Kristyna Krulova, Jan Hodek, Ales Zabransky, Ivan Hirsch, and Jan Weber. 2021. "ATM-Dependent Phosphorylation of Hepatitis B Core Protein in Response to Genotoxic Stress" Viruses 13, no. 12: 2438. https://doi.org/10.3390/v13122438
APA StyleLubyova, B., Tikalova, E., Krulova, K., Hodek, J., Zabransky, A., Hirsch, I., & Weber, J. (2021). ATM-Dependent Phosphorylation of Hepatitis B Core Protein in Response to Genotoxic Stress. Viruses, 13(12), 2438. https://doi.org/10.3390/v13122438