
SARS-CoV-2 ORF3a Induces Incomplete Autophagy via the Unfolded Protein Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Plasmids and Transfection
2.3. Antibodies
2.4. Western Blotting
2.5. Confocal Immunofluorescence Microscopy
2.6. RNA Isolation and Quantitative Real-Time PCR
2.7. Statistical Analysis
3. Results
3.1. SARS-CoV-2 ORF3a Promoted the Expression of Autophagy-Related Genes
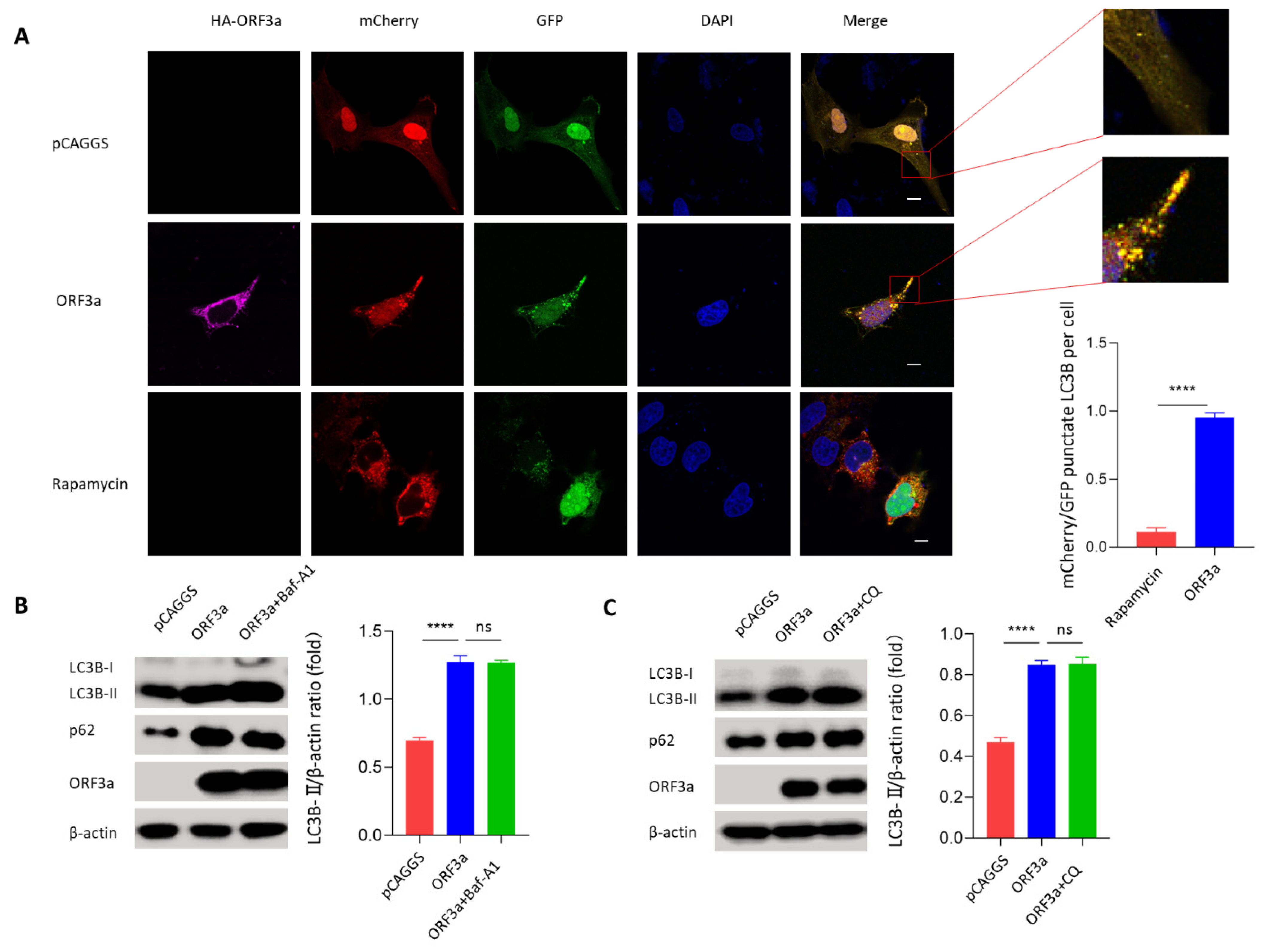
3.2. ORF3a Could Induce Autophagy
3.3. ORF3a-Induced Autophagy Was an Incomplete Process
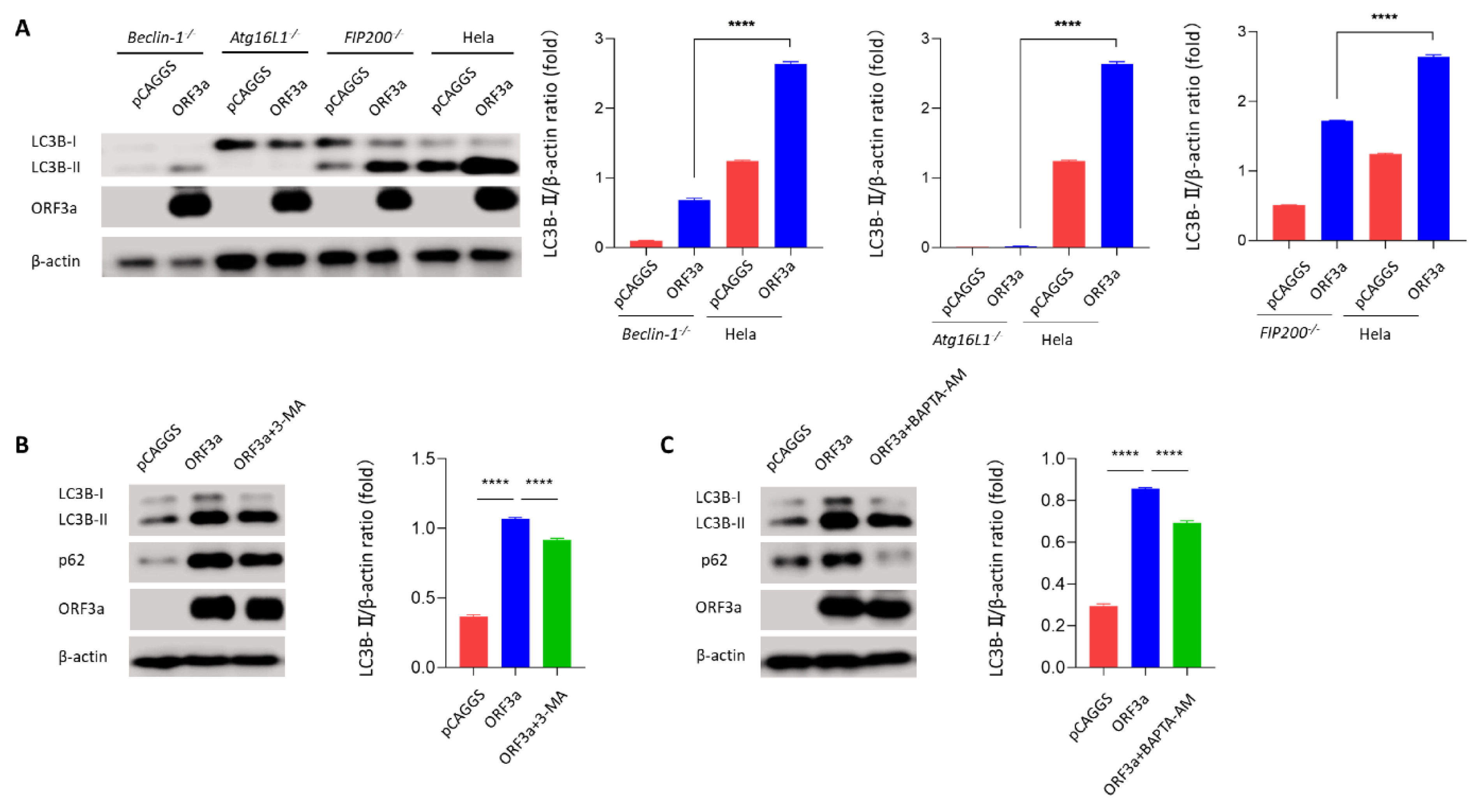
3.4. ORF3a-Induced Autophagy Was Dependent on the Classical Autophagy
3.5. ORF3a Induced Autophagy through Unfolded Protein Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matsuzawa-Ishimoto, Y.; Hwang, S.; Cadwell, K. Autophagy and Inflammation. Annu. Rev. Immunol. 2018, 36, 73–101. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Munz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V.; Greber, U.F. The endoplasmic reticulum unfolded protein response—Homeostasis, cell death and evolution in virus infections. FEMS Microbiol. Rev. 2021, 45, fuab016. [Google Scholar] [CrossRef]
- Hwang, J.W.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Frakes, A.E.; Dillin, A. The UPR(ER): Sensor and Coordinator of Organismal Homeostasis. Mol. Cell 2017, 66, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- B’chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2 alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [Green Version]
- Song, S.L.; Tan, J.; Miao, Y.Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef] [PubMed]
- Deegan, S.; Saveljeva, S.; Gorman, A.M.; Samali, A. Stress-induced self-cannibalism: On the regulation of autophagy by endoplasmic reticulum stress. Cell. Mol. Life Sci. 2013, 70, 2425–2441. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.; McGrath, M.E.; Hu, Z.Q.; Ariannejad, S.; Weston, S.; Frieman, M.; Jackson, W.T. Coronavirus interactions with the cellular autophagy machinery. Autophagy 2020, 16, 2131–2139. [Google Scholar] [CrossRef]
- Dogra, P.; Ruiz-Ramirez, J.; Sinha, K.; Butner, J.D.; Pelaez, M.J.; Rawat, M.; Yellepeddi, V.K.; Pasqualini, R.; Arap, W.; Sostman, H.D.; et al. Innate Immunity Plays a Key Role in Controlling Viral Load in COVID-19: Mechanistic Insights from a Whole-Body Infection Dynamics Model. ACS Pharmacol. Transl. 2021, 4, 248–265. [Google Scholar] [CrossRef]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 73, 529–557. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.X.; Fung, T.S.; Chong, K.K.L.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014, 109, 97–109. [Google Scholar] [CrossRef]
- Korner, R.W.; Majjouti, M.; Alcazar, M.A.A.; Mahabir, E. Of Mice and Men: The Coronavirus MHV and Mouse Models as a Translational Approach to Understand SARS-CoV-2. Viruses 2020, 12, 880. [Google Scholar] [CrossRef]
- Tan, Y.J. The Severe Acute Respiratory Syndrome (SARS)-coronavirus 3a protein may function as a modulator of the trafficking properties of the spike protein. Virol. J. 2005, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Castano-Rodriguez, C.; Honrubia, J.M.; Gutierrez-Alvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Baguena, C.; Queralt-Martin, M.; et al. Role of Severe Acute Respiratory Syndrome Coronavirus Viroporins E, 3a, and 8a in Replication and Pathogenesis. mBio 2018, 9, e02325-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, R.; Yang, R.F.; Shi, M.D.; Jiang, M.R.; Xie, Y.H.; Ruan, H.Q.; Jiang, X.S.; Shi, L.; Zhou, H.; Zhang, L.; et al. Characterization of the 3a protein of SARS-associated coronavirus in infected Vero e6 cells and SARS patients. J. Mol. Biol. 2004, 341, 271–279. [Google Scholar] [CrossRef]
- Zhang, J.; Cruz-Cosme, R.; Zhuang, M.W.; Liu, D.X.; Liu, Y.A.; Teng, S.L.; Wang, P.H.; Tang, Q.Y. A systemic and molecular study of subcellular localization of SARS-CoV-2 proteins. Signal Transduct. Target. Ther. 2020, 5, 269. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.M.; Sorum, B.; Mali, S.S.; Hoel, C.M.; Sridharan, S.; Remis, J.P.; Toso, D.B.; Kotecha, A.; Bautista, D.M.; Brohawn, S.G. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Sun, H.; Pei, R.J.; Mao, B.L.; Zhao, Z.Y.; Li, H.H.; Lin, Y.; Lu, K.F. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2021, 56, 427–442.e5. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Savova, A.; Danieli, A.; Romanov, J.; Tremel, S.; Ebner, M.; Peterbauer, T.; Sztacho, M.; Trapannone, R.; Tarafder, A.K.; et al. p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 2018, 37, e98308. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanasios, E.; Walker, S.A.; Okkenhaug, H.; Manifava, M.; Hummel, E.; Zimmermann, H.; Ahmed, Q.; Domart, M.C.; Collinson, L.; Ktistakis, N.T. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat. Commun. 2016, 7, 12420. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Coleman, O.I.; Haller, D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 2019, 10, 2825. [Google Scholar] [CrossRef]
- Waye, M.M.Y.; Law, P.T.W.; Wong, C.H.; Au, T.C.C.; Chuck, C.P.; Kong, S.K.; Chan, P.K.S.; To, K.F.; Lo, A.W.I.; Chan, J.Y.W.; et al. The 3a protein of SARS-coronavirus induces apoptosis in Vero E6 cells. In Proceedings of the IEEE Engineering in Medicine and Biology 27th Annual Conference, Shanghai, China, 17–18 January 2006; pp. 7482–7485. [Google Scholar]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The SARS Coronavirus 3a Protein Causes Endoplasmic Reticulum Stress and Induces Ligand-Independent Downregulation of the Type 1 Interferon Receptor. PLoS ONE 2009, 4, e8342. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.F.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.D.; Shen, H.M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef]
- Pasquier, B. Autophagy inhibitors. Cell. Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Cao, R.Y.; Zhang, L.K.; Yang, X.L.; Liu, J.; Xu, M.Y.; Shi, Z.L.; Hu, Z.H.; Zhong, W.; Xiao, G.F. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Batista, R. Using Chloroquine and Hydroxychloroquine in the Treatment of COVID-19: Does It Make Sense? Indian J. Pharm. Educ. 2020, 54, 865–874. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.F.; Zhu, Y.X.; Lin, X.; Ren, C.W.; Zhao, J.C.; Wang, F.F.; Gao, X.C.; Xiao, R.; Zhao, L.Z.; Chen, H.C.; et al. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy 2019, 15, 1163–1181. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Leli, N.M.; Koumenis, C.; Amaravadi, R.K. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin. Cancer Biol. 2020, 66, 116–128. [Google Scholar] [CrossRef]
- Chan, C.P.; Siu, K.L.; Chin, K.T.; Yuen, K.Y.; Zheng, B.; Jin, D.Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [Green Version]
- Siu, K.L.; Chan, C.P.; Kok, K.H.; Woo, P.C.; Jin, D.Y. Comparative analysis of the activation of unfolded protein response by spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus HKU1. Cell Biosci. 2014, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Rashid, F.; Dzakah, E.E.; Wang, H.Y.; Tang, S.X. The ORF8 protein of SARS-CoV-2 induced endoplasmic reticulum stress and mediated immune evasion by antagonizing production of interferon beta. Virus Res. 2021, 296, 198350. [Google Scholar] [CrossRef]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Santerre, M.; Arjona, S.P.; Allen, C.N.S.; Shcherbik, N.; Sawaya, B.E. Why do SARS-CoV-2 NSPs rush to the ER? J. Neurol. 2021, 268, 2013–2022. [Google Scholar] [CrossRef]
- Axten, J.M.; Medina, J.R.; Feng, Y.H.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-Methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a Potent and Selective First-in-Class Inhibitor of Protein Kinase R (PKR)-like Endoplasmic Reticulum Kinase (PERK). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef]
- Guirao-Abad, J.P.; Weichert, M.; Albee, A.; Deck, K.; Askew, D.S. A Human IRE1 Inhibitor Blocks the Unfolded Protein Response in the Pathogenic Fungus Aspergillus fumigatus and Suggests Noncanonical Functions within the Pathway. mSphere 2020, 5, e00879-20. [Google Scholar] [CrossRef]
- Yang, S.; Zhu, J.; Zhou, X.; Wang, H.; Li, X.; Zhao, A. Induction of the unfolded protein response (UPR) during pseudorabies virus infection. Vet. Microbiol. 2019, 239, 108485. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Xie, W.W.; Yin, D.Y.; Luo, R.; Liu, M.; Guo, F.J. ATG5 and ATG7 induced autophagy interplays with UPR via PERK signaling. Cell Commun. Signal. 2019, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Song, X.X.; Lee, D.H.; Dilly, A.K.; Lee, Y.S.; Choudry, H.A.; Kwon, Y.T.; Bartlett, D.L.; Lee, Y.J. Crosstalk Between Apoptosis and Autophagy Is Regulated by the Arginylated BiP/Beclin-1/p62 Complex. Mol. Cancer Res. 2018, 16, 1077–1091. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, W.-q.; Yu, X.-j.; Zhou, C.-m. SARS-CoV-2 ORF3a Induces Incomplete Autophagy via the Unfolded Protein Response. Viruses 2021, 13, 2467. https://doi.org/10.3390/v13122467
Su W-q, Yu X-j, Zhou C-m. SARS-CoV-2 ORF3a Induces Incomplete Autophagy via the Unfolded Protein Response. Viruses. 2021; 13(12):2467. https://doi.org/10.3390/v13122467
Chicago/Turabian StyleSu, Wen-qing, Xue-jie Yu, and Chuan-min Zhou. 2021. "SARS-CoV-2 ORF3a Induces Incomplete Autophagy via the Unfolded Protein Response" Viruses 13, no. 12: 2467. https://doi.org/10.3390/v13122467
APA StyleSu, W. -q., Yu, X. -j., & Zhou, C. -m. (2021). SARS-CoV-2 ORF3a Induces Incomplete Autophagy via the Unfolded Protein Response. Viruses, 13(12), 2467. https://doi.org/10.3390/v13122467