
Genetic and Antigenic Characterization of Avian Avulavirus Type 6 (AAvV-6) Circulating in Canadian Wild Birds (2005–2017)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and RNA Extraction
2.2. Virus Isolation
2.3. Hemagglutination Inhibition (HI) Assay
2.4. Sequencing
2.5. Phylogenetic Analysis
2.6. Development of a Duplex Fusion Gene Based Real-Time RT-PCR for AMPV-6
2.7. Fusion Real-Time RT-PCR Conditions
2.8. Biological Characterization of AAvV-6 Viruses in Chickens
2.9. Serology
2.10. Antigenic Cartography
3. Results
3.1. Virus Isolation and Characterization
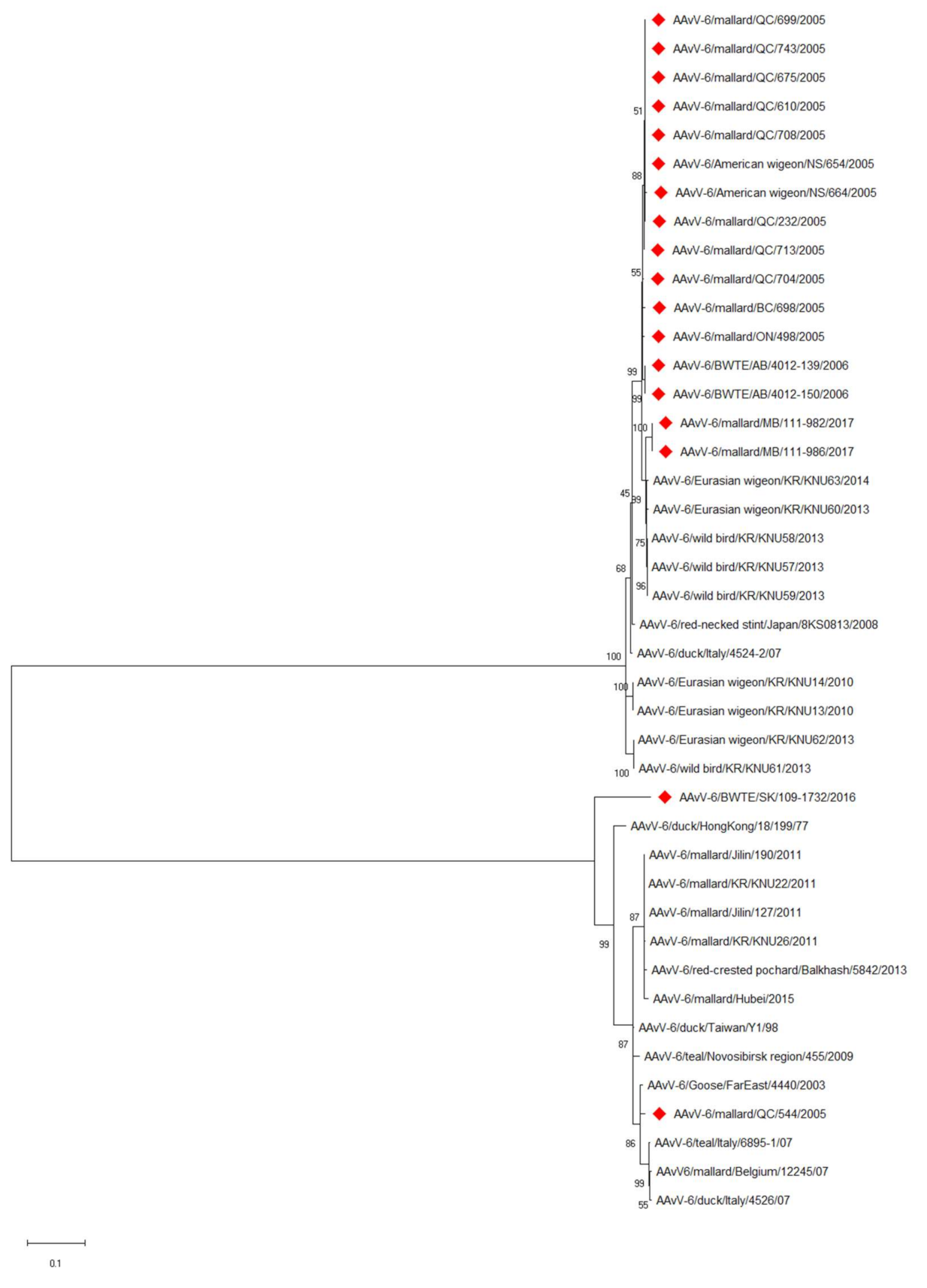
3.2. Molecular and Phylogenetic Characterization
3.3. Development of a Duplex Fusion Gene Based Real-Time RT-PCR for AAvV-6
3.4. Biological Characterisation AAvV-6 Viruses in SPF Chickens
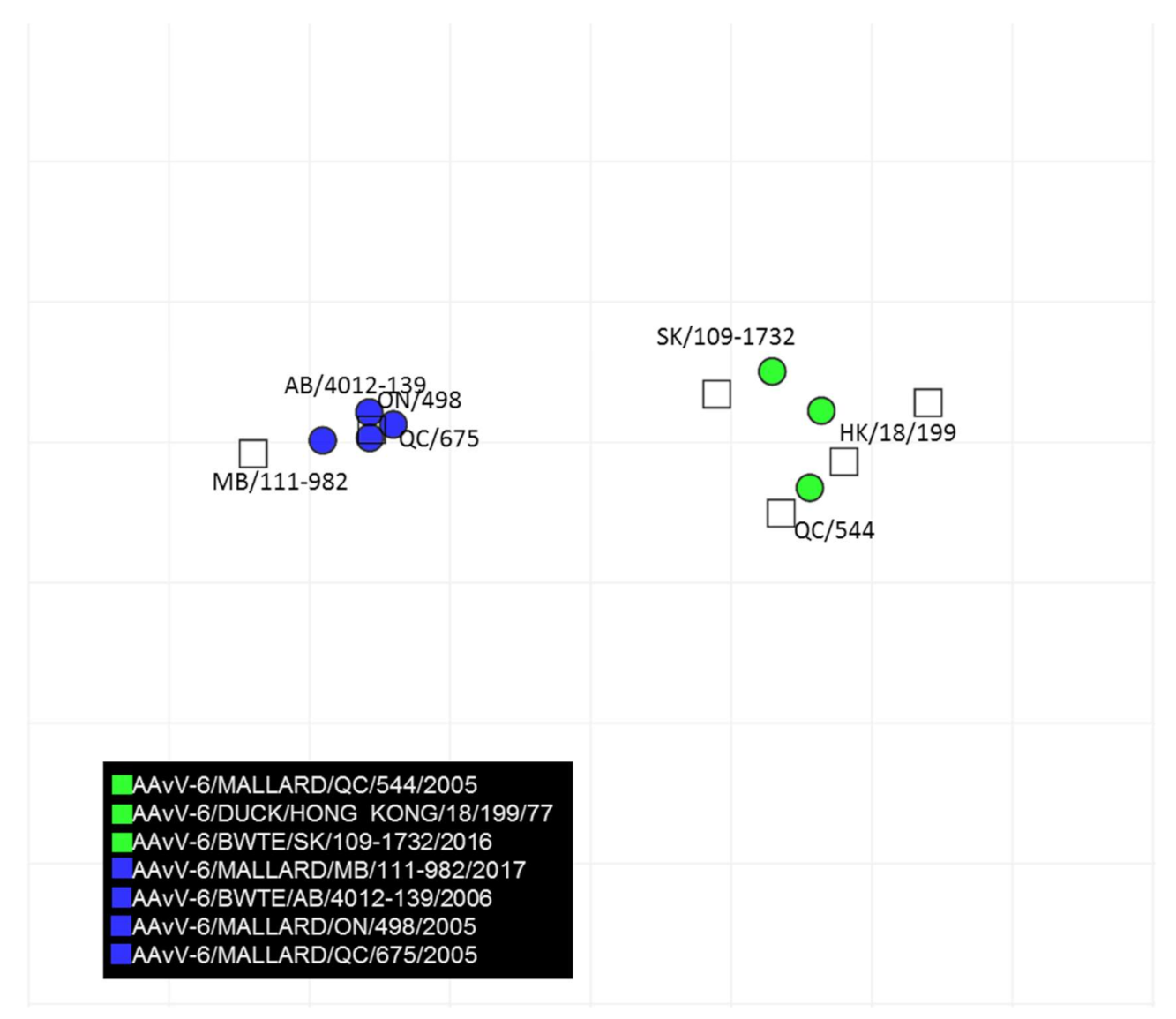
3.5. Antigenic Cartography
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexander, D. Avian paramyxoviruses 2–9. In Diseases of Poultry, 11th ed.; Saif, Y.M., Ed.; Iowa State University Press: Ames, IA, USA, 2003; pp. 88–92. [Google Scholar]
- Chang, P.C.; Hsieh, M.L.; Shien, J.H.; Graham, D.A.; Lee, M.S.; Shieh, H.K. Complete nucleotide sequence of avian paramyxovirus type 6 isolated from ducks. J. Gen. Virol. 2001, 82, 2157–2168. [Google Scholar] [CrossRef]
- Morrison, T.; McQuain, C.; McGinnes, L. Complementation between avirulent Newcastle disease virus and a fusion protein gene expressed from a retrovirus vector: Requirements for membrane fusion. J. Virol. 1991, 65, 813–822. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Kielian, M.; Helenius, A. Membrane fusion proteins of enveloped animal viruses. Q. Rev. Biophys. 1983, 16, 151–195. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Subbiah, M.; Samuel, A.S.; Collins, P.L.; Samal, S.K. Roles of the fusion and hemagluttinin-neuraminidase proteins in replication, tropism and pathogenicity of avian paramyxoviruses. J. Virol. 2011, 85, 8582–8596. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.J.; Afonso, C.L.; Spackman, E.; Scott, M.A.; Pedersen, J.C.; Senne, D.A.; Brown, J.D.; Fuller, C.M.; Uhart, M.M.; Karesh, W.B.; et al. Evidence for a new avian paramyxovirus serotype 10 detected in Rockhopper penguins from the Falkland Islands. J. Virol. 2010, 84, 11496–11504. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses. Virus Taxonomy: 2018b Release EC 51, Berlin, Germany. 20 July 2019. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/negative-sense-rna-viruses/w/paramyxoviridae/1193/genus-orthoavulavirus (accessed on 20 January 2021).
- Shortridge, K.F.; Alexander, D.J.; Collins, M.S. Isolation and properties of viruses from poultry in Hong Kong which represents a new (sixth) distinct group of avian paramyxoviruses. J. Gen. Virol. 1980, 49, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Subbiah, M.; Kumar, S.; De Nardi, R.; Terregino, C.; Collins, P.L.; Samal, S.K. Complete genome sequences of avian paramyxovirus serotype 6 prototype strain Hong Kong and a recent novel strain from Italy: Evidence for the existence of subgroups within the serotype. Virus Res. 2010, 150, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Shi-Hee, K.; Xiao, S.; Shive, H.; Collins, P.L.; Samal, S.K. Replication, neurotropism and pathogenecity of avian paramyxovirus serotypes 1-9 in chickens and ducks. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Tian, Z.; Chai, H.; Li, F.; Sun, J.; Chen, G.; Hu, X.; Xiang, W. Complete nucleotide sequence of avian paramyxovirus type 6 strain JL isolated from mallard ducks in China. J. Virol. 2012, 86, 13112. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, I.A.; Sharsov, K.; Yurchenko, K.; Korneev, D.; Glushchenko, A.; Alikina, T.; Kabilov, M.; Bi, Y.; Liu, W.; Gubanova, N.; et al. Characterization of avian paramyxovirus type 6 isolated from a Eurasian teal in the intersection of migratory flyways in Russia. Arch. Virol. 2016, 161, 3275–3279. [Google Scholar] [CrossRef]
- Choi, K.S.; Kim, J.Y.; Lee, H.J.; Jang, M.J.; Kwon, H.M.; Sung, H.W. Genetic diversity of avian paramyxovirus type 6 isolated from wild ducks in the Republic of Korea. J. Wildl. Dis. 2018, 54, 558–563. [Google Scholar] [CrossRef]
- Chen, Y.; Ding, Z.; Liu, X.; Chen, J.; Li, J.; Fei, Y.; Liu, Z.; Soteger, T.; Bi, Y.; Yin, R. Biological and phylogenetic characterization of a novel hemagglutination-negative avian avulavirus 6 isolated from wild waterfowl in China. Transbound. Emerg. Dis. 2018, 65, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Warke, A.; Stallknecht, D.; Williams, S.M.; Pritchard, N.; Mundt, E. Comparative study on the pathogenicity and immunogenicity of wild bird isolates of avian paramyxovirus 2, 4, and 6 in chickens. Avian Pathol. 2008, 37, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Pasick, J.; Berhane, Y.; Kehler, H.; Hisanaga, T.; Handel, K.; Robinson, J.; Ojkic, D.; Kibenge, F.; Fortin, M.; King, R.; et al. Survey of influenza A viruses circulating in wild birds in Canada 2005–2007. Avian Dis. 2010, 54, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Parmley, E.J.; Bastien, N.; Booth, T.F.; Bowes, V.; Buck, P.A.; Breault, A.; Caswell, D.; Daoust, P.Y.; Davies, J.C.; Elahi, S.M.; et al. Wild bird influenza survey, Canada. 2005. Emerg. Infect. Dis. 2008, 4, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Parmley, E.J.; Soos, C.; Breault, A.; Leighton, F.A.; Fortin, M.; Jenkins, E.; Kibenge, F.; King, R.; McAloney, K.; Pasick, J.; et al. Detection of low pathogenic avian influenza viruses in wild ducks from Canada: Comparison of two sampling methods. J. Wildl. Dis. 2011, 47, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Weingartl, H.; Berhane, Y.; Hisanaga, T.; Neufeld, J.; Kehler, H.; Embury-Hyatt, C.; Hooper-McGrevy, K.; Kasloff, S.; Dalman, B.; Bystrom, J.; et al. Genetic and pathobiologic characterization of pandemic H1N1 2009 influenza viruses from a naturally infected swine herd. J. Virol. 2010, 84, 2245–2256. [Google Scholar] [CrossRef]
- Webster, R.G.; Cox, N.; Stöhr, K. WHO Manual on Animal Influenza Diagnosis and Surveillance. 2002. Available online: http://www.who.int/csr/resources/publications/influenza/whocdscsrncs20025rev.pdf (accessed on 15 November 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetic analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Lapedes, A.S.; De Jong, J.C.; Bestebroer, T.M.; Rimmelzwaan, G.F.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Mapping the antigenic and genetic evolution of influenza virus. Science 2004, 305, 371–376. [Google Scholar] [CrossRef] [PubMed]
- De Jong, J.C.; Smith, D.J.; Lapedes, A.S.; Donatelli, I.; Campitelli, L.; Barigazzi, G.; Van Reeth, K.; Jones, T.C.; Rimmelzwaan, G.F.; Osterhaus, A.D.; et al. Antigenic and genetic evolution of swine influenza A (H3N2) viruses in Europe. J. Virol. 2007, 81, 4315–4322. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.L.; Gandham, R.K.; Subbiah, M. Molecular evolution and genetic variations of V and W proteins derived by RNA editing in Avian Paramyxoviruses. Sci. Rep. 2020, 10, 9532. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.S.; Bashiruddin, J.B.; Alexander, D.J. Deduced amino acid sequences at the fusion protein cleavage site of Newcastle disease virus: Evidence for the existence of a new genus within the subfamily Paramyxovirinae. J. Gen. Virol. 1993, 80, 131–136. [Google Scholar]
- Diel, D.G.; Da Silva, L.H.; Liu, H.; Wang, Z.; Miller, P.J.; Afonso, C.L. Genetic diversity of avian paramyxovirus type 1: Proposal for a unified nomenclature and classification system of Newcastle disease virus genotypes. Infect. Genet. Evol. 2012, 12, 1770–1779. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer and Probe | Genotype | Position |
|---|---|---|
| Forward 1 (Genotype-1) | 3′-CACCCTTAAYCGAATTTTTACACC-5′ | 252–275 |
| Forward 2 (Genotype-2) | 3′-CACATTGAACCGCATATTCACRCC-5′ | 252–275 |
| Rev. 1–2 (Genotype type-1 & 2) | 3′-GCYCTTAACCARGCCCAGGA-5′ | 442–443 |
| Probe (Genotype-1) | CALFluorOrange560-CAACCAGAACCCTGCTCCAG-BHQ1 | 327–346 |
| Probe (Genotype-2) | FAM-CTCACCTCACTCCATACGTG-BHQ1 |
| Virus | Genotype | Antisera | |||||
|---|---|---|---|---|---|---|---|
| Duck/HongKong/18/199/77 | BWTE/SK/109-1732/2016 | Mallard/QC/675/2005 | |||||
| Ck#1 | Ck#2 | Ck#483 | Ck#485 | Ck#602 | Ck#604 | ||
| Mallard/QC/544/2005 | 1 | 512 | 512 | 32 | 16 | 8 | 16 |
| Duck/Hong Kong/18/199/77 | 1 | 512 | 1024 | 64 | 16 | 8 | 16 |
| BWTE/SK/109-1732/2016 | 1 | 256 | 512 | 64 | 32 | 8 | 16 |
| Mallard/MB/111-982/2017 | 2 | 128 | 64 | 4 | 4 | 64 | 128 |
| BWTE/AB/4012-139/2006 | 2 | 128 | 32 | 4 | 4 | 128 | 128 |
| Mallard/ON/498/2005 | 2 | 64 | 64 | 8 | 4 | 64 | 128 |
| Mallard/QC/675/2005 | 2 | 128 | 64 | 8 | 4 | 64 | 128 |
| Virus | Length | Location (Province) | Year | Genotype | Cleavage Site | HA Titre | HI Titre | Genbank Accession Number |
|---|---|---|---|---|---|---|---|---|
| AAvV-6 mallard/QC/544/2005 | 16,236 | Quebec | 2005 | G1 | PAPEPR*LVGA | 4096 | 128 | MW338846 |
| AAvV-6 BWTE/SK/OTH109-1732/2016 | 16,236 | Saskatchewan | 2016 | G1 | PAPEPR*LVGA | 1024 | 64 | MW338847 |
| AAvV-6 mallard/AB/4012-139/2006 | 16,230 | Alberta | 2006 | G2 | SIREPR*LIGA | 256 | 8 | MW338858 |
| AAvV-6 mallard/AB/4012-150/2006 | 16,230 | Alberta | 2006 | G2 | SIREPR*LIGA | 64 | 16 | MW338859 |
| AAvV-6 mallard/QC/232/2005 | 16,230 | Quebec | 2005 | G2 | SIREPR*LIGA | 256 | 4 | MW338849 |
| AAvV-6 mallard/QC/610/2005 | 16,216 a | Quebec | 2005 | G2 | SIREPR*LIGA | 256 | 4 | MW338850 |
| AAvV-6 mallard/QC/675/2005 | 16,230 | Quebec | 2005 | G2 | SIREPR*LIGA | 1024 | 8 | MW338851 |
| AAvV-6 mallard/QC/699/2005 | 16,230 | Quebec | 2005 | G2 | SIREPR*LIGA | 512 | 16 | MW338852 |
| AAvV-6 mallard/QC/708/2005 | 16,230 | Quebec | 2005 | G2 | SIREPR*LIGA | 512 | 8 | MW338853 |
| AAvV-6 mallard/QC/713/2005 | 1638 b | Quebec | 2005 | G2 | REPR*LIGA | 2048 | 8 | MW338862 |
| AAvV-6 mallard/QC/743/2005 | 16,230 | Quebec | 2005 | G2 | SIREPR*LIGA | 2048 | 4 | MW338854 |
| AAvV-6 mallard/BC/698/2005 | 16,230 | British Colombia | 2005 | G2 | SIREPR*LIGA | 32 | 4 | MW338856 |
| AAvV-6 mallard/BC/704/2005 | 16,230 | British Colombia | 2005 | G2 | SIREPR*LIGA | 256 | 4 | MW338857 |
| AAvV-6 American wigeon/NS/654/2005 | 16,230 | Nova Scotia | 2005 | G2 | SIREPR*LIGA | 512 | 16 | MW338848 |
| AAvV-6 American wigeon/NS/664/2005 | 1638 b | Nova Scotia | 2005 | G2 | SIREPR*LIGA | 128 | 16 | MW338863 |
| AAVV-6 mallard/ON/498/2005 | 16,230 | Ontario | 2005 | G2 | SIREPR*LIGA | 64 | 8 | MW338855 |
| AAvV-6 mallard/MB/OTH111-982/2017 | 16,230 | Manitoba | 2017 | G2 | SIREPR*LIGA | 512 | 8 | MW338860 |
| AAvV-6 mallard/MB/OTH111-986/2017 | 16,230 | Manitoba | 2017 | G2 | SIREPR*LIGA | did not hemaggluttinate | MW338861 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hisanaga, T.; Soos, C.; Lewis, N.; Lung, O.; Suderman, M.; Berhane, Y. Genetic and Antigenic Characterization of Avian Avulavirus Type 6 (AAvV-6) Circulating in Canadian Wild Birds (2005–2017). Viruses 2021, 13, 543. https://doi.org/10.3390/v13040543
Hisanaga T, Soos C, Lewis N, Lung O, Suderman M, Berhane Y. Genetic and Antigenic Characterization of Avian Avulavirus Type 6 (AAvV-6) Circulating in Canadian Wild Birds (2005–2017). Viruses. 2021; 13(4):543. https://doi.org/10.3390/v13040543
Chicago/Turabian StyleHisanaga, Tamiko, Catherine Soos, Nicola Lewis, Oliver Lung, Matthew Suderman, and Yohannes Berhane. 2021. "Genetic and Antigenic Characterization of Avian Avulavirus Type 6 (AAvV-6) Circulating in Canadian Wild Birds (2005–2017)" Viruses 13, no. 4: 543. https://doi.org/10.3390/v13040543
APA StyleHisanaga, T., Soos, C., Lewis, N., Lung, O., Suderman, M., & Berhane, Y. (2021). Genetic and Antigenic Characterization of Avian Avulavirus Type 6 (AAvV-6) Circulating in Canadian Wild Birds (2005–2017). Viruses, 13(4), 543. https://doi.org/10.3390/v13040543