
Mitigating Future Respiratory Virus Pandemics: New Threats and Approaches to Consider


Abstract
:1. Introduction
1.1. Human Metapneumovirus (hMPV)
1.2. Rhinovirus Group C (HRV-C)
1.3. Enterovirus A71 (EV-A71)
1.4. Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV)
1.5. Human Adenovirus 14 Strain (HAdV14)
1.6. 2009 H1N1 Influenza A Pandemic (H1N1pdm09) Virus
1.7. Middle East Respiratory Syndrome Coronavirus (MERS-CoV)
1.8. Enterovirus D68 (EV-D68)
1.9. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
2. Economic Costs of Emerging Infectious Diseases
3. Predicting Novel Pathogen Emergence
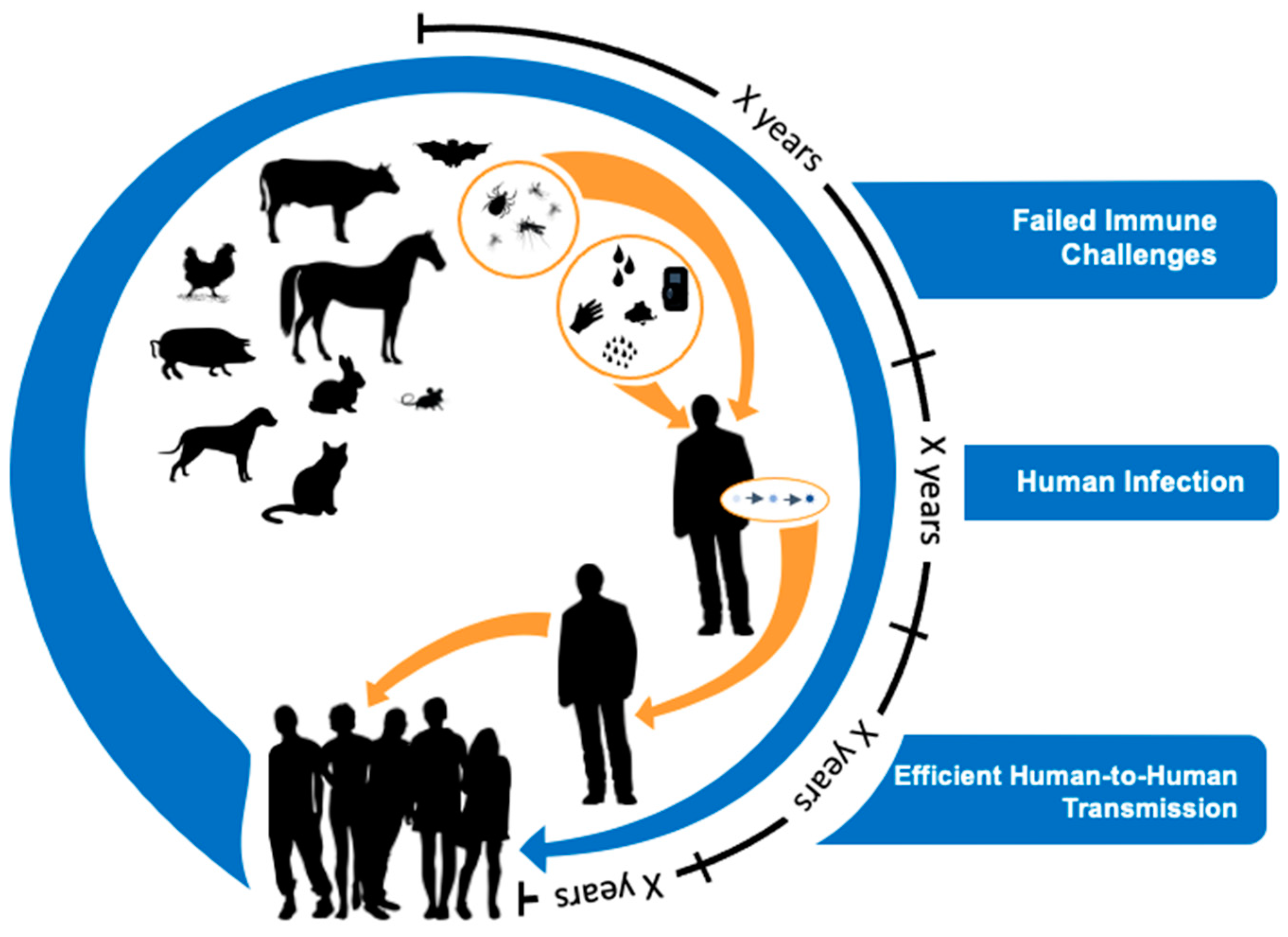
4. Virus Spillover to Humans Is Common, but Most Viruses Are Poorly Adapted to Humans
5. Evolutionary Perspectives on Efficient Human-to-Human Transmission
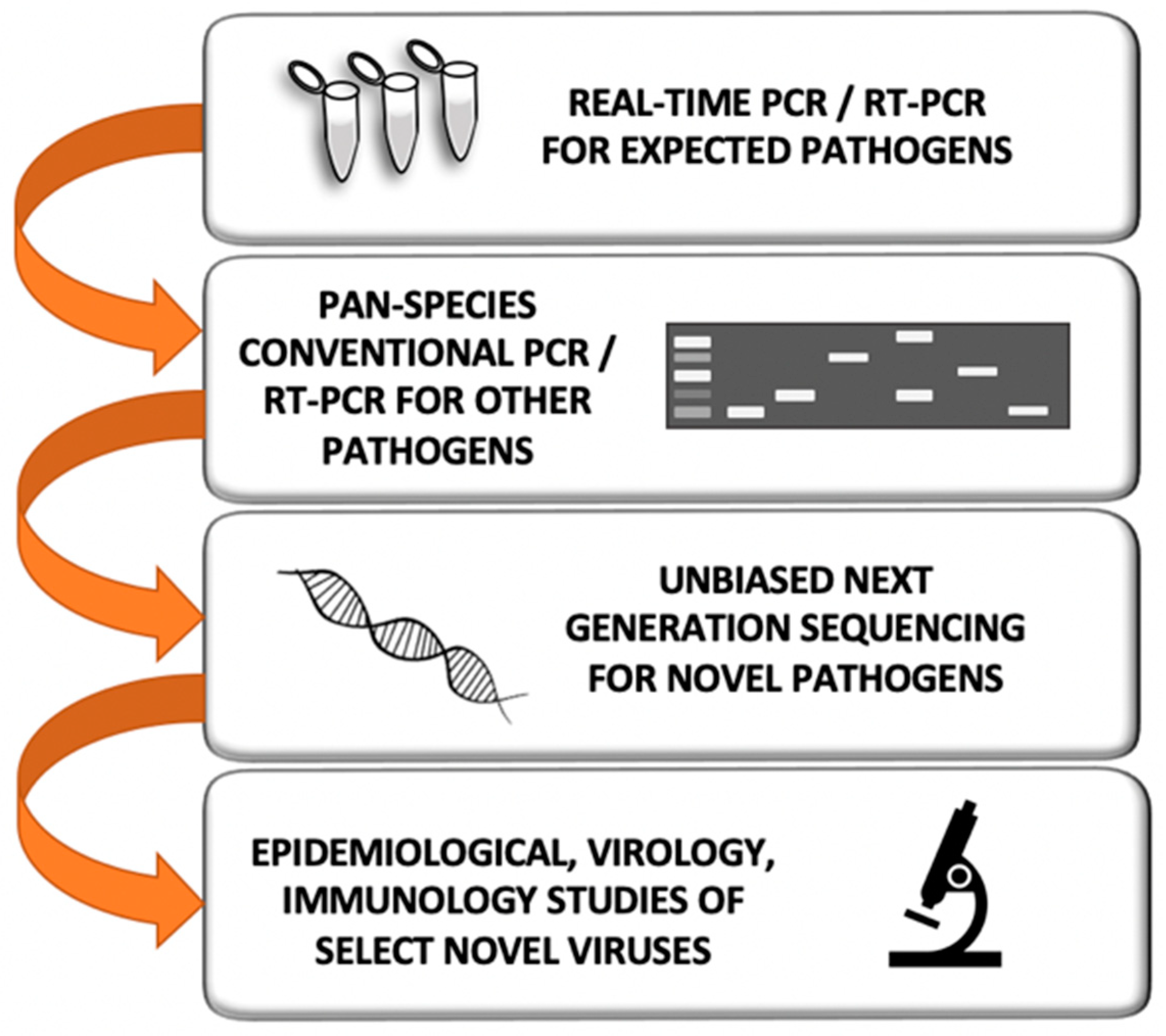
6. How Do We Currently Search for Novel Prepandemic Viruses?
7. What Would Be the Best Strategy to Detect Novel Prepandemic Viruses
7.1. Among Which Animals Is Prevalence of Respiratory Viruses High?
7.2. Where Is the Animal Reservoir-Human Contact Rate High?
7.3. From Which Viruses and Viral Hosts Is the Probability of Zoonotic Virus Transmission to Humans Highest?
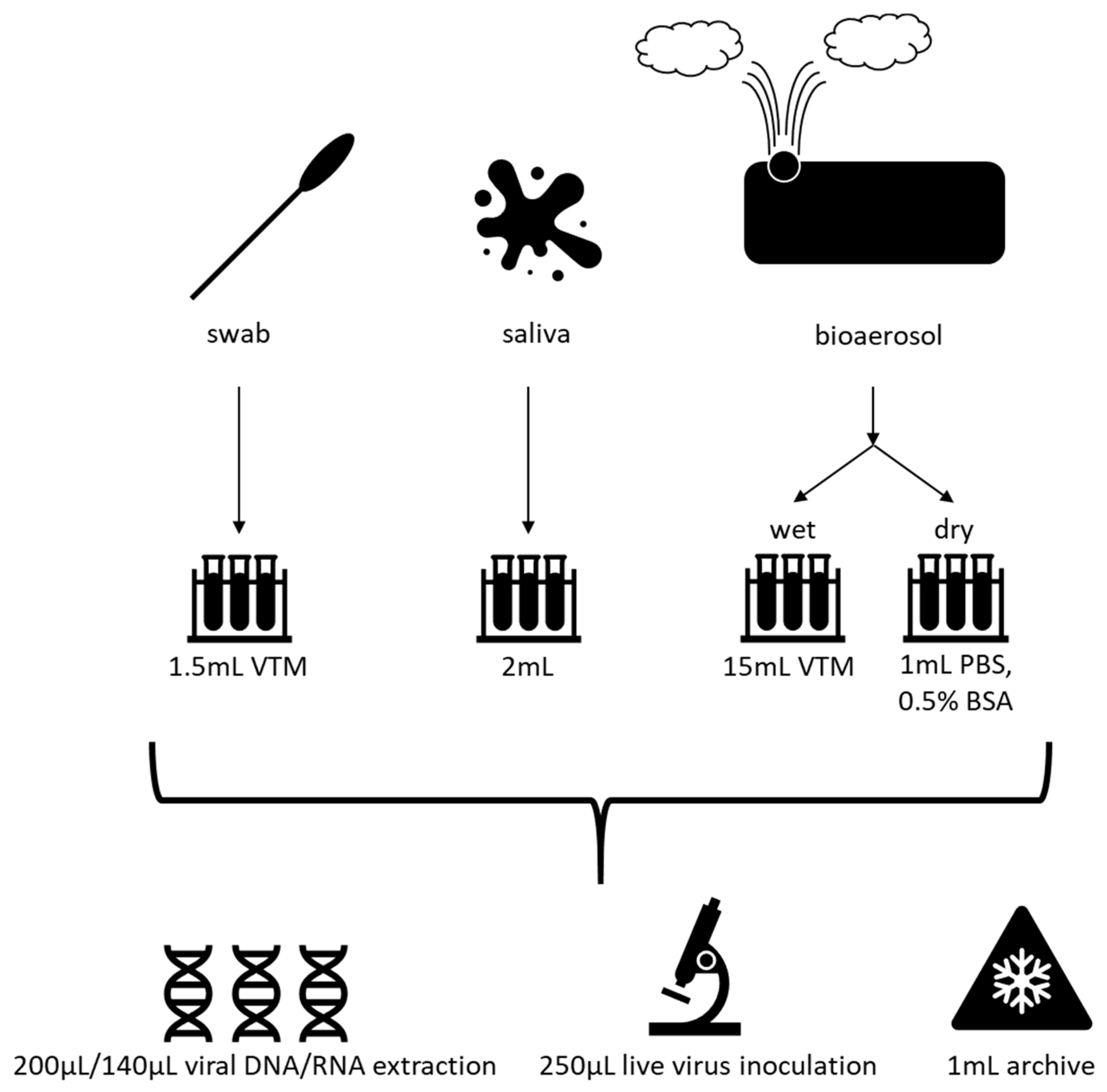
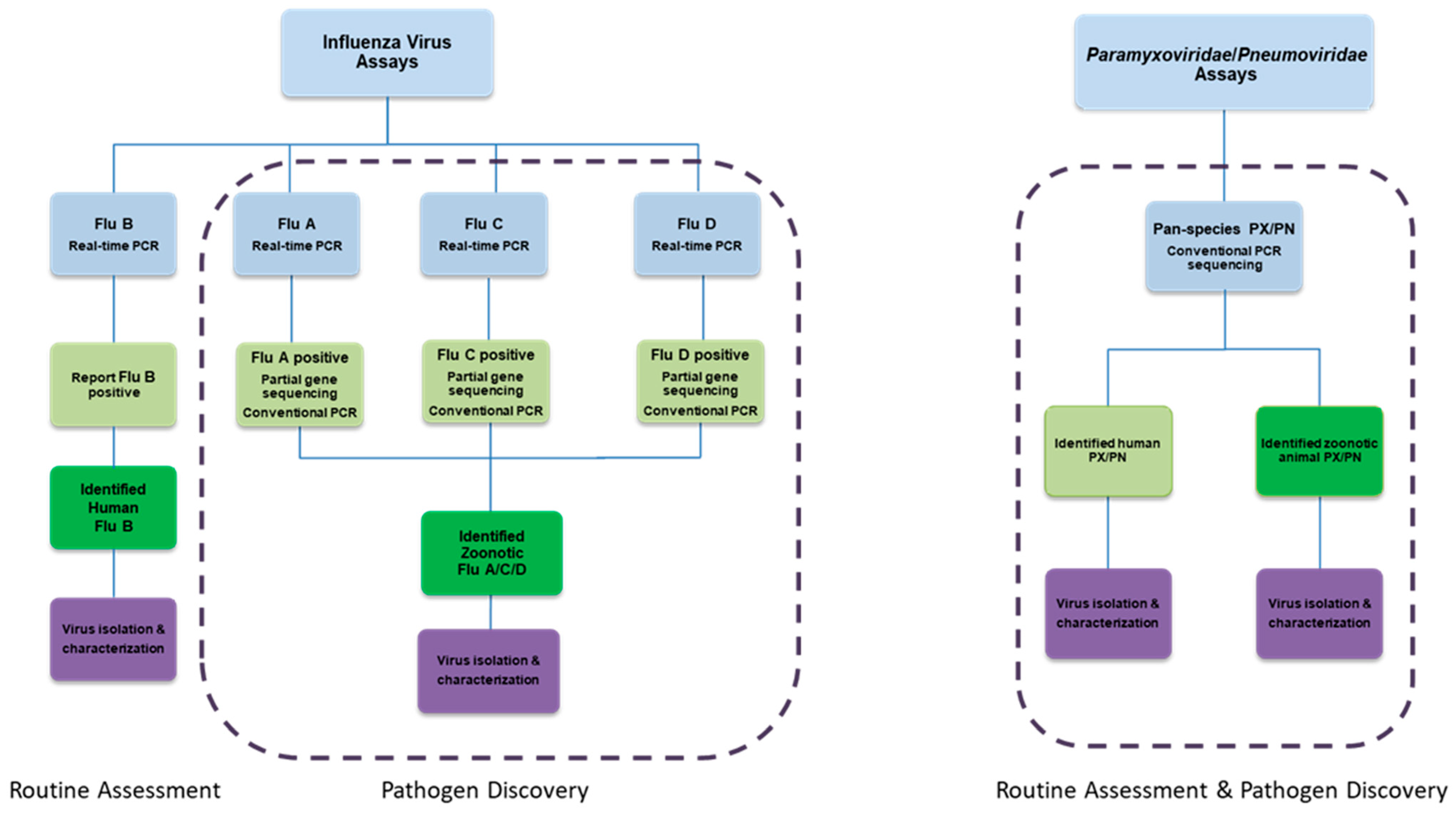
8. Efficient Field Collection, Clinical and Laboratory Methods
9. Novel Virus Risk Assessments
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haynes, A.K.; Fowlkes, A.L.; Schneider, E.; Mutuc, J.D.; Armstrong, G.L.; Gerber, S.I. Human Metapneumovirus Circulation in the United States, 2008 to 2014. Pediatrics 2016, 137, e20152927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.; Yip, C.; Woo, P.; Yuen, K.-Y. Human rhinovirus C: A newly discovered human rhinovirus species. Emerg. Health Threat. J. 2010, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Puenpa, J.; Wanlapakorn, N.; Vongpunsawad, S.; Poovorawan, Y. The History of Enterovirus A71 Outbreaks and Molecular Epidemiology in the Asia-Pacific Region. J. Biomed. Sci. 2019, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention SARS Basics Fact Sheet. Available online: https://www.cdc.gov/sars/about/fs-sars.html (accessed on 26 February 2021).
- Gautret, P.; Gray, G.C.; Charrel, R.N.; Odezulu, N.G.; Al-Tawfiq, J.A.; Zumla, A.; Memish, Z.A. Emerging viral respiratory tract infections—environmental risk factors and transmission. Lancet Infect. Dis. 2014, 14, 1113–1122. [Google Scholar] [CrossRef]
- Shrestha, S.S.; Swerdlow, D.L.; Borse, R.H.; Prabhu, V.S.; Finelli, L.; Atkins, C.Y.; Owusu-Edusei, K.; Bell, B.; Mead, P.S.; Biggerstaff, M.; et al. Estimating the Burden of 2009 Pandemic Influenza A (H1N1) in the United States (April 2009–April 2010). Clin. Infect. Dis. 2010, 52, S75–S82. [Google Scholar] [CrossRef] [Green Version]
- WHO Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Available online: http://www.who.int/emergencies/mers-cov/en/ (accessed on 22 September 2020).
- Holm-Hansen, C.C.; Midgley, S.E.; Fischer, T.K. Global emergence of enterovirus D68: A systematic review. Lancet Infect. Dis. 2016, 16, e64–e75. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention Enterovirus D68. Available online: https://www.cdc.gov/non-polio-enterovirus/about/ev-d68.html (accessed on 27 February 2020).
- World Health Organization Coronavirus Disease 2019 (COVID-19) Situation Report—36. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/ (accessed on 26 February 2021).
- Hoogen, B.G.V.D.; De Jong, J.C.; Groen, J.; Kuiken, T.; De Groot, R.; Fouchier, R.A.; Osterhaus, A.D. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 2001, 7, 719–724. [Google Scholar] [CrossRef]
- Williams, J.V.; Wang, C.K.; Yang, C.; Tollefson, S.J.; House, F.S.; Heck, J.M.; Chu, M.; Brown, J.B.; Lintao, L.D.; Quinto, J.D.; et al. The Role of Human Metapneumovirus in Upper Respiratory Tract Infections in Children: A 20-Year Experience. J. Infect. Dis. 2006, 193, 387–395. [Google Scholar] [CrossRef]
- Schildgen, V.; Hoogen, B.V.D.; Fouchier, R.; Tripp, R.A.; Alvarez, R.; Manoha, C.; Williams, J. Human Metapneumovirus: Lessons Learned over the First Decade. Clin. Microbiol. Rev. 2011, 24, 734–754. [Google Scholar] [CrossRef] [Green Version]
- Shafagati, N.; Williams, J. Human metapneumovirus—What we know now. F1000Research 2018, 7, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayali, G.; Ortiz, E.J.; Chorazy, M.L.; Nagaraja, K.V.; DeBeauchamp, J.; Webby, R.J.; Gray, G.C. Serologic Evidence of Avian Metapneumovirus Infection Among Adults Occupationally Exposed to Turkeys. Vector Borne Zoonotic Dis. 2011, 11, 1453–1458. [Google Scholar] [CrossRef]
- Velayudhan, B.T.; Nagaraja, K.V.; Thachil, A.J.; Shaw, D.P.; Gray, G.C.; Halvorson, D.A. Human Metapneumovirus in Turkey Poults. Emerg. Infect. Dis. 2006, 12, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Negrey, J.D.; Reddy, R.B.; Scully, E.J.; Phillips-Garcia, S.; Owens, L.A.; Langergraber, K.E.; Mitani, J.C.; Thompson, M.E.; Wrangham, R.W.; Muller, M.N.; et al. Simultaneous outbreaks of respiratory disease in wild chimpanzees caused by distinct viruses of human origin. Emerg. Microbes Infect. 2019, 8, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamson, D.; Renwick, N.; Kapoor, V.; Liu, Z.; Palacios, G.; Ju, J.; Dean, A.; George, K.S.; Briese, T.; Lipkin, W.I. MassTag Polymerase-Chain-Reaction Detection of Respiratory Pathogens, Including a New Rhinovirus Genotype, That Caused Influenza-Like Illness in New York State during 2004–2005. J. Infect. Dis. 2006, 194, 1398–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, E.J.; Basnet, S.; Wrangham, R.W.; Muller, M.N.; Otali, E.; Hyeroba, D.; Grindle, K.A.; Pappas, T.E.; Thompson, M.E.; Machanda, Z.; et al. Lethal Respiratory Disease Associated with Human Rhinovirus C in Wild Chimpanzees, Uganda, 2013. Emerg. Infect. Dis. 2018, 24, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention Notes from the Field: Enterovirus A71 Neurologic Disease in Children—Colorado, 2018. Morb. Mortal. Wkly. Rep. 2018, 67, 1017. [CrossRef] [Green Version]
- Fieldhouse, J.K.; Wang, X.; Mallinson, K.A.; Tsao, R.W.; Gray, G.C. A systematic review of evidence that enteroviruses may be zoonotic. Emerg. Microbes Infect. 2018, 7, 164. [Google Scholar] [CrossRef] [Green Version]
- Luk, H.K.; Li, X.; Fung, J.; Lau, S.K.; Woo, P.C. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infect. Genet. Evol. 2019, 71, 21–30. [Google Scholar] [CrossRef]
- Borkenhagen, L.K.; Fieldhouse, J.K.; Seto, D.; Gray, G.C. Are adenoviruses zoonotic? A systematic review of the evidence. Emerg. Microbes Infect. 2019, 8, 1679–1687. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention. The Burden of the Influenza A H1N1pdm09 Virus since the 2009 Pandemic. Available online: https://www.cdc.gov/flu/pandemic-resources/burden-of-h1n1.html (accessed on 26 February 2021).
- Group, W.B. One Health Operational Framework for Strengthening Human, Animal, and Environmental Public Health Systems at Their Interface; The World Bank: Washington, DC, USA, 2018. [Google Scholar]
- Joo, H.; Maskery, B.A.; Berro, A.D.; Rotz, L.D.; Lee, Y.K.; Brown, C.M. Economic Impact of the 2015 MERS Outbreak on the Republic of Korea’s Tourism-Related Industries. Health Secur. 2019, 17, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Fuller, T.L.; Gilbert, M.; Martin, V.; Cappelle, J.; Hosseini, P.; Njabo, K.Y.; Aziz, S.A.; Xiao, X.; Daszak, P.; Smith, T.B. Predicting Hotspots for Influenza Virus Reassortment. Emerg. Infect. Dis. 2013, 19, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nat. Cell Biol. 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.; Murray, K.A.; Zambrana-Torrelio, C.; Morse, S.S.; Rondinini, C.; Di Marco, M.; Breit, N.; Olival, K.J.; Daszak, P. Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 2017, 8, 1124. [Google Scholar] [CrossRef]
- Li, X.-L.; Yang, Y.; Sun, Y.; Chen, W.-J.; Sun, R.-X.; Liu, K.; Ma, M.-J.; Liang, S.; Yao, H.-W.; Gray, G.C.; et al. Risk Distribution of Human Infections with Avian Influenza H7N9 and H5N1 virus in China. Sci. Rep. 2015, 5, 18610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Ma, J.; Lai, A.; Gray, G.C.; Su, S.; Li, S. Avian influenza A(H7N9) virus and mixed live poultry-animal markets in Guangdong province: A perfect storm in the making? Emerg. Microbes Infect. 2015, 4, e63. [Google Scholar] [CrossRef]
- Fang, L.-Q.; Li, X.-L.; Liu, K.; Li, Y.-J.; Yao, H.-W.; Liang, S.; Yang, Y.; Feng, Z.-J.; Gray, G.C.; Cao, W.-C. Mapping Spread and Risk of Avian Influenza A (H7N9) in China. Sci. Rep. 2013, 3, 2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Bi, Y.; Wong, G.; Gray, G.C.; Gao, G.F.; Li, S. Epidemiology, Evolution, and Recent Outbreaks of Avian Influenza Virus in China. J. Virol. 2015, 89, 8671–8676. [Google Scholar] [CrossRef] [Green Version]
- Crespi, B. Evolutionary medical insights into the SARS-CoV-2 pandemic. Evol. Med. Public Health 2020, 2020, 314–322. [Google Scholar] [CrossRef]
- Gibb, R.; Redding, D.W.; Chin, K.Q.; Donnelly, C.A.; Blackburn, T.M.; Newbold, T.; Jones, K.E. Zoonotic host diversity increases in human-dominated ecosystems. Nat. Cell Biol. 2020, 584, 398–402. [Google Scholar] [CrossRef]
- Plowright, R.K.; Eby, P.; Hudson, P.J.; Smith, I.L.; Westcott, D.; Bryden, W.L.; Middleton, D.; Reid, P.A.; McFarlane, R.A.; Martin, G.; et al. Ecological dynamics of emerging bat virus spillover. Proc. R. Soc. B Boil. Sci. 2015, 282, 20142124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, M.K.; Becker, D.J.; Peel, A.J.; Justice, N.V.; Lunn, T.; Crowley, D.E.; Jones, D.N.; Eby, P.; Sánchez, C.A.; Plowright, R.K. Changing resource landscapes and spillover of henipaviruses. Ann. N. Y. Acad. Sci. 2018, 1429, 78–99. [Google Scholar] [CrossRef]
- Longdon, B.; Hadfield, J.D.; Webster, C.L.; Obbard, D.J.; Jiggins, F.M. Host Phylogeny Determines Viral Persistence and Replication in Novel Hosts. PLoS Pathog. 2011, 7, e1002260. [Google Scholar] [CrossRef] [Green Version]
- Streicker, D.G.; Turmelle, A.S.; Vonhof, M.J.; Kuzmin, I.V.; McCracken, G.F.; Rupprecht, C.E. Host Phylogeny Constrains Cross-Species Emergence and Establishment of Rabies Virus in Bats. Science 2010, 329, 676–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, N.; Griffin, R.; Franz, M.; Omotayo, M.; Nunn, C.L. Phylogenetic host specificity and understanding parasite sharing in primates. Ecol. Lett. 2012, 15, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Köndgen, S.; Calvignac-Spencer, S.; Grützmacher, K.; Keil, V.; Mätz-Rensing, K.; Nowak, K.; Metzger, S.; Kiyang, J.; Lübke-Becker, A.; Deschner, T.; et al. Author Correction: Evidence for Human Streptococcus pneumoniae in wild and captive chimpanzees: A potential threat to wild populations. Sci. Rep. 2020, 10, 9062, Erratum in 2017, 7, 14581. [Google Scholar] [CrossRef]
- Köndgen, S.; Kühl, H.; N’Goran, P.K.; Walsh, P.D.; Schenk, S.; Ernst, N.; Biek, R.; Formenty, P.; Mätz-Rensing, K.; Schweiger, B.; et al. Pandemic Human Viruses Cause Decline of Endangered Great Apes. Curr. Biol. 2008, 18, 260–264. [Google Scholar] [CrossRef] [Green Version]
- Antia, R.; Regoes, R.R.; Koella, J.C.; Bergstrom, C.T. The role of evolution in the emergence of infectious diseases. Nat. Cell Biol. 2003, 426, 658–661. [Google Scholar] [CrossRef]
- Wallace, B. Can “Stepping Stones” Form Stairways? Am. Nat. 1989, 133, 578–579. [Google Scholar] [CrossRef]
- Sikkema, R.; Reina, S.; Freidl, G.S.; De Bruin, E.; Koopmans, M. Weighing serological evidence of human exposure to animal influenza viruses—A literature review. Eurosurveillance 2016, 21, 30388. [Google Scholar] [CrossRef] [Green Version]
- Borkenhagen, L.K.; Wang, G.-L.; Simmons, R.A.; Bi, Z.-Q.; Lu, B.; Wang, X.-J.; Wang, C.-X.; Chen, S.-H.; Song, S.-X.; Li, M.; et al. High Risk of Influenza Virus Infection Among Swine Workers: Examining a Dynamic Cohort in China. Clin. Infect. Dis. 2020, 71, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Decaestecker, E.; Gaba, S.; Raeymaekers, J.A.; Stoks, R.; Van Kerckhoven, L.; Ebert, D.; De Meester, L. Host-parasite ‘Red Queen’ dynamics archived in pond sediment. Nature 2007, 450, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Lively, C.M. Antagonistic Coevolution and Sex. Evol. Educ. Outreach 2010, 3, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Ewald, P.W. The evolution of virulence: A unifying link between parasitology and ecology. J. Parasitol. 1995, 81, 659–669. [Google Scholar] [CrossRef]
- Alizon, S.; Hurford, A.; Mideo, N.; Van Baalen, M. Virulence evolution and the trade-off hypothesis: History, current state of affairs and the future. J. Evol. Biol. 2008, 22, 245–259. [Google Scholar] [CrossRef]
- Fraser, C.; Hollingsworth, T.D.; Chapman, R.; De Wolf, F.; Hanage, W.P. Variation in HIV-1 set-point viral load: Epidemiological analysis and an evolutionary hypothesis. Proc. Natl. Acad. Sci. USA 2007, 104, 17441–17446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Commission on Strengthening America’s Health Security; Center for Strategic International Studies. Biological Threats to U.S. National Security: Testimony before the Senate Armed Services Subcommittee on Emerging Threats and Capabilities. Available online: https://healthsecurity.csis.org/events/biological-threats-to-u-s-national-security-testimony-before-the-senate-armed-services-subcommittee-on-emerging-threats-and-capabilities/ (accessed on 1 November 2020).
- Holmes, E.C.; Rambaut, A.; Andersen, K.G. Pandemics: Spend on surveillance, not prediction. Nat. Cell Biol. 2018, 558, 180–182. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Holmes, E.C. Predicting virus emergence amid evolutionary noise. Open Biol. 2017, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Yong, E. Is It Possible to Predict the Next Pandemic Science? Available online: https://www.theatlantic.com/science/archive/2017/10/pandemic-prediction-challenge/543954/ (accessed on 1 November 2020).
- Morrison, J. Can Virus Hunters Stop the Next Pandemic before It Happens? Available online: https://www.smithsonianmag.com/science-nature/how-to-stop-next-animal-borne-pandemic-180967908/ (accessed on 1 November 2020).
- Robbins, J. Before the Next Pandemic, an Ambitious Push to Catalog Viruses in Wildlife. Available online: https://e360.yale.edu/features/before-the-next-pandemic-an-ambitious-push-to-catalog-viruses-in-wildlife (accessed on 26 February 2021).
- U.S. Agency for International Development. Appendix I Discovery & Exploration of Emerging Pathogens—Viral Zoonoses (DEEP VZN). Available online: https://govtribe.com/opportunity/federal-grant-opportunity/discovery-exploration-of-emerging-pathogens-viral-zoonoses-deep-vzn-7200aa21rfa00005 (accessed on 26 February 2021).
- Graham, B. Issue 15: COVID-19: A Wake-up Call for More Fundamental Research in Immunology. Available online: https://www.humanvaccinesproject.org/covid-post/issue-15-covid-19-a-wake-up-call-for-more-fundamental-research-in-immunology/ (accessed on 1 November 2020).
- Graham, B.S.; Corbett, K.S. Prototype pathogen approach for pandemic preparedness: World on fire. J. Clin. Investig. 2020, 130, 3348–3349. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.S.; Sullivan, N.J. Emerging viral diseases from a vaccinology perspective: Preparing for the next pandemic. Nat. Immunol. 2017, 19, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.D.; Ma, M.-J.; Wang, G.L.; Bi, Z.-Q.; Lu, B.; Wang, X.-J.; Wang, C.-X.; Chen, S.-H.; Qian, Y.-H.; Song, S.-X.; et al. Prospective surveillance for influenza A virus in Chinese swine farms. Emerg. Microbes Infect. 2018, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rwego, I.B.; Babalobi, O.O.; Musotsi, P.; Nzietchueng, S.; Tiambo, C.K.; Kabasa, J.D.; Naigaga, I.; Kalema-Zikusoka, G.; Pelican, K. One Health capacity building in sub-Saharan Africa. Infect. Ecol. Epidemiol. 2016, 6, 75. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, L.; Wang, G.; Lu, J. One Health in China. Infect. Ecol. Epidemiol. 2016, 6, 33843. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.S.; Dahal, R.; Kakkar, M.; Debnath, N.; Rahman, M.; Dorjee, S.; Naeem, K.; Wijayathilaka, T.; Sharma, B.K.; Maidanwal, N.; et al. One Health research and training and government support for One Health in South Asia. Infect. Ecol. Epidemiol. 2016, 6, 33842. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.A.; McKenzie, J.; Woldeyohannes, S.M. One Health research and training in Australia and New Zealand. Infect. Ecol. Epidemiol. 2016, 6, 33799. [Google Scholar] [CrossRef]
- Sikkema, R.; Koopmans, M. One Health training and research activities in Western Europe. Infect. Ecol. Epidemiol. 2016, 6, 33703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroud, C.; Kaplan, B.; Logan, J.E.; Gray, G.C. One Health training, research, and outreach in North America. Infect. Ecol. Epidemiol. 2016, 6, 33680. [Google Scholar] [CrossRef]
- Sinclair, J. Importance of a One Health approach in advancing global health security and the Sustainable Development Goals. Rev. Sci. Tech. OIE 2019, 38, 145–154. [Google Scholar] [CrossRef]
- O’Brien, M.K.; Perez, A.M.; Errecaborde, K.M. Transforming the One Health workforce: Lessons learned from initiatives in Africa, Asia and Latin America. Rev. Sci. Tech. 2019, 38, 239–250. [Google Scholar] [CrossRef]
- Bright-Ponte, S.; Walters, B.; Tate, H.; Durso, L.; Whichard, J.; Bjork, K.; Shivley, C.; Beaudoin, A.; Cook, K.; Thacker, E.; et al. One Health and antimicrobial resistance, a United States perspective. Rev. Sci. Tech. OIE 2019, 38, 173–184. [Google Scholar] [CrossRef]
- Bird, B.H.; Mazet, J.A. Detection of Emerging Zoonotic Pathogens: An Integrated One Health Approach. Annu. Rev. Anim. Biosci. 2018, 6, 121–139. [Google Scholar] [CrossRef] [Green Version]
- Degeling, C.; Johnson, J.; Kerridge, I.; Wilson, A.; Ward, M.R.; Stewart, C.; Gilbert, G.L. Implementing a One Health approach to emerging infectious disease: Reflections on the socio-political, ethical and legal dimensions. BMC Public Health 2015, 15, 1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiu, L.; Binder, R.A.; Alarja, N.A.; Kochek, K.; Coleman, K.K.; Than, S.T.; Bailey, E.S.; Bui, V.N.; Toh, T.-H.; Erdman, D.D.; et al. A RT-PCR assay for the detection of coronaviruses from four genera. J. Clin. Virol. 2020, 128, 104391. [Google Scholar] [CrossRef] [PubMed]
- Fieldhouse, J.K.; Bailey, E.S.; Toh, T.-H.; Hii, K.-C.; Mallinson, K.A.; Ting, J.; Lednicky, J.A.; Berita, A.; Nguyen, T.T.; Galan, D.; et al. Panspecies molecular assays detect viral pathogens missed by real-time PCR/reverse-transcriptase PCR among pneumonia patients, Sarawak, Malaysia. Trop. Dis. Travel Med. Vaccines 2020, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.S.; Mullis, L.B.; Pereira, O.; Saif, L.J.; Vlasova, A.; Zhang, X.; Owens, R.J.; Paulson, D.; Taylor, D.; Haynes, L.M.; et al. Human Respiratory Coronaviruses Detected In Patients with Influenza-Like Illness in Arkansas, USA. Virol. Mycol. 2014, S2, 004. [Google Scholar] [CrossRef]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Elbadry, M.A.; Alam, M.M.; Stephenson, C.J.; Bonny, T.S.; Loeb, J.C.; Telisma, T.; Chavannes, S.; et al. Emergence of porcine delta-coronavirus pathogenic infections among children in Haiti through independent zoonoses and convergent evolution. medRxiv 2021. [Google Scholar]
- Bailey, E.S.; Fieldhouse, J.K.; Alarja, N.A.; Chen, D.D.; Kovalik, M.E.; Zemke, J.N.; Choi, J.Y.; Borkenhagen, L.K.; Toh, T.-H.; Lee, J.S.Y.; et al. First sequence of influenza D virus identified in poultry farm bioaerosols in Sarawak, Malaysia. Trop. Dis. Travel Med. Vaccines 2020, 6, 1–4. [Google Scholar] [CrossRef]
- Bailey, E.S.; Borkenhagen, L.K.; Choi, J.Y.; Greer, A.E.; Culhane, M.R.; Gray, G.C. A feasibility study of conducting surveillance for swine pathogens in slurry from North Carolina swine farms. Sci. Rep. 2020, 10, 1–6. [Google Scholar] [CrossRef]
- Yondon, M.; Zayat, B.; Nelson, M.I.; Heil, G.L.; Anderson, B.D.; Lin, X.; Halpin, R.A.; McKenzie, P.P.; White, S.K.; Wentworth, D.E.; et al. Equine Influenza A(H3N8) Virus Isolated from Bactrian Camel, Mongolia. Emerg. Infect. Dis. 2014, 20, 2144–2147. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Smith, J.O.; George, D.; Pepin, K.M.; Pitzer, V.E.; Pulliam, J.R.C.; Dobson, A.P.; Hudson, P.J.; Grenfell, B.T. Epidemic Dynamics at the Human-Animal Interface. Science 2009, 326, 1362–1367. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Sheng, Z.; Huang, C.; Wang, D.; Li, F. Influenza D virus. Curr. Opin. Virol. 2020, 44, 154–161. [Google Scholar] [CrossRef]
- Kenney, S.P.; Wang, Q.; Vlasova, A.; Jung, K.; Saif, L. Naturally Occurring Animal Coronaviruses as Models for Studying Highly Pathogenic Human Coronaviral Disease. Veter. Pathol. 2020, 300985820980842. [Google Scholar] [CrossRef]
- Gray, G.C.; Merchant, J.A. Pigs, pathogens, and public health. Lancet Infect. Dis. 2018, 18, 372–373. [Google Scholar] [CrossRef]
- Myers, K.P.; Olsen, C.W.; Setterquist, S.F.; Capuano, A.W.; Donham, K.J.; Thacker, E.L.; Merchant, J.A.; Gray, G.C. Are Swine Workers in the United States at Increased Risk of Infection with Zoonotic Influenza Virus? Clin. Infect. Dis. 2006, 42, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.P.; Leibler, J.H.; Price, L.B.; Otte, J.M.; Pfeiffer, D.U.; Tiensin, T.; Silbergeld, E.K. The Animal-Human Interface and Infectious Disease in Industrial Food Animal Production: Rethinking Biosecurity and Biocontainment. Public Health Rep. 2008, 123, 282–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawal, O.U.; Fraqueza, M.J.; Bouchami, O.; Worning, P.; Bartels, M.D.; Gonçalves, M.L.; Paixão, P.; Gonçalves, E.; Toscano, C.; Empel, J.; et al. Foodborne Origin and Local and Global Spread of Staphylococcus saprophyticus Causing Human Urinary Tract Infections. Emerg. Infect. Dis. 2021, 27, 880–893. [Google Scholar] [CrossRef]
- Freidl, G.S.; Meijer, A.; De Bruin, E.; De Nardi, M.; Munoz, O.; Capua, I.; Breed, A.C.; Harris, K.; Hill, A.; Kosmider, R.; et al. Influenza at the animal–human interface: A review of the literature for virological evidence of human infection with swine or avian influenza viruses other than A(H5N1). Eurosurveillance 2014, 19, 20793. [Google Scholar] [CrossRef] [Green Version]
- Borkenhagen, L.K.; Salman, M.D.; Ma, M.-J.; Gray, G.C. Animal influenza virus infections in humans: A commentary. Int. J. Infect. Dis. 2019, 88, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Vlasova, A.N.; Kenney, S.P.; Saif, L.J. Emerging and re-emerging coronaviruses in pigs. Curr. Opin. Virol. 2019, 34, 39–49. [Google Scholar] [CrossRef]
- Wang, X.; Bailey, E.S.; Qi, X.; Yu, H.; Bao, C.; Gray, G.C. Bioaerosol Sampling at a Live Animal Market in China: A Noninvasive Approach for Detecting Emergent Viruses. Open Forum Infect. Dis. 2019, ofaa134. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, L.; Eckard, L.; Epperson, W.B.; Long, L.-P.; Smith, D.; Huston, C.; Genova, S.; Webby, R.; Wan, X.-F. Influenza D virus infection in Mississippi beef cattle. Virology 2015, 486, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hause, B.M.; Ducatez, M.; Collin, E.A.; Ran, Z.; Liu, R.; Sheng, Z.; Armien, A.; Kaplan, B.; Chakravarty, S.; Hoppe, A.D.; et al. Isolation of a Novel Swine Influenza Virus from Oklahoma in 2011 Which Is Distantly Related to Human Influenza C Viruses. PLoS Pathog. 2013, 9, e1003176. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Pabbaraju, K.; Wong, S.; Wong, A.; May-Hadford, J.; Tellier, R.; Fonseca, K. Detection of influenza C virus by a real-time RT-PCR assay. Influ. Other Respir. Viruses 2013, 7, 954–960. [Google Scholar] [CrossRef] [Green Version]
- Van Boheemen, S.; Bestebroer, T.; Verhagen, J.; Osterhaus, A.; Pas, S.; Herfst, S.; Fouchier, R. A Family-Wide RT-PCR Assay for Detection of Paramyxoviruses and Application to a Large-Scale Surveillance Study. PLoS ONE 2012, 7, e34961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Sridhar, S.; Lau, S.K.P.; Liu, J.; Ou, J.; Yan, Y.; Zhao, S.; Lan, W.; Guan, W.; Wu, J.; et al. Molecular Typing of Human Respiratory Adenoviruses with Universal PCR and Sequencing Primers for Three Major Capsid Genes: Penton base, Hexon, and Fiber. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Wellehan, J.F.X.; Johnson, A.J.; Harrach, B.; Benkö, M.; Pessier, A.P.; Johnson, C.M.; Garner, M.M.; Childress, A.; Jacobson, E.R. Detection and Analysis of Six Lizard Adenoviruses by Consensus Primer PCR Provides Further Evidence of a Reptilian Origin for the Atadenoviruses. J. Virol. 2004, 78, 13366–13369. [Google Scholar] [CrossRef] [Green Version]
- Oberste, M.S.; Feeroz, M.M.; Maher, K.; Nix, W.A.; Engel, G.A.; Hasan, K.M.; Begum, S.; Oh, G.; Chowdhury, A.H.; Pallansch, M.A.; et al. Characterizing the Picornavirus Landscape among Synanthropic Nonhuman Primates in Bangladesh, 2007 to 2008. J. Virol. 2012, 87, 558–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bil-Lula, I.; De Franceschi, N.; Pawlik, K.; Mieczyslaw, W. Improved real-time PCR assay for detection and quantification of all 54 known types of human adenoviruses in clinical samples. Med. Sci. Monit. 2012, 18, 221–228. [Google Scholar]
- World Health Organization. Centers for Disease Control and Prevention Enterovirus Surveillance Guidelines. Available online: https://www.euro.who.int/__data/assets/pdf_file/0020/272810/EnterovirusSurveillanceGuidelines.pdf (accessed on 1 November 2020).
- Loens, K.; Van Loon, A.M.; Coenjaerts, F.E.J.; Van Aarle, Y.; Goossens, H.; Wallace, P.S.; Claas, E.J.C.; Ieven, M.; On Behalf of the GRACE Study Group. Performance of Different Mono- and Multiplex Nucleic Acid Amplification Tests on a Multipathogen External Quality Assessment Panel. J. Clin. Microbiol. 2011, 50, 977–987. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention CDC 2019-Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel; Centers for Disease Control and Prevention, Division of Viral Diseases: Atlanta, GA USA. Available online: https://www.fda.gov/media/134922/download (accessed on 25 March 2021).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Years | Virus | Geographical Range | Estimated Morbidity (Infections) | Estimated Mortality (Deaths) |
|---|---|---|---|---|
| 1995+ | Human metapneumovirus † [1] | Worldwide | 42,464 | At least 41 (Table S1) |
| 1995+ | Rhinovirus Group C † [2] | Worldwide | 1632 | At least 7 (Table S2) |
| 1998+ | Enterovirus A71 [3] | Worldwide | 12.8 M * | At least 3747 * |
| 2003–2004 | SARS-CoV † [4] | 29 countries | 8098 | 774 |
| 2006+ | Adenovirus 14 [5] | United States, Ireland, China, Canada | >750 | At least 13 |
| 2009+ | influenza A (H1N1)pdm09 virus † [6] | Worldwide | 100.5 M ‡ | 75,000 ‡ |
| 2012+ | MERS-CoV † [7] | 27 countries | 2494 § | 858 § |
| 2014+ | Enterovirus D68 [8,9] | Worldwide | 2529 ¶ | At least 17 ¶ |
| 2019+ | SARS-CoV-2 † [10] | Worldwide | >120,000,000 as of 03/15/2021 | >2,659,000 as of 03/15/2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gray, G.C.; Robie, E.R.; Studstill, C.J.; Nunn, C.L. Mitigating Future Respiratory Virus Pandemics: New Threats and Approaches to Consider. Viruses 2021, 13, 637. https://doi.org/10.3390/v13040637
Gray GC, Robie ER, Studstill CJ, Nunn CL. Mitigating Future Respiratory Virus Pandemics: New Threats and Approaches to Consider. Viruses. 2021; 13(4):637. https://doi.org/10.3390/v13040637
Chicago/Turabian StyleGray, Gregory C., Emily R. Robie, Caleb J. Studstill, and Charles L. Nunn. 2021. "Mitigating Future Respiratory Virus Pandemics: New Threats and Approaches to Consider" Viruses 13, no. 4: 637. https://doi.org/10.3390/v13040637
APA StyleGray, G. C., Robie, E. R., Studstill, C. J., & Nunn, C. L. (2021). Mitigating Future Respiratory Virus Pandemics: New Threats and Approaches to Consider. Viruses, 13(4), 637. https://doi.org/10.3390/v13040637