
The Phylogeography of Potato Virus X Shows the Fingerprints of Its Human Vector

 ,
,  , ,
, ,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Virus Isolates
2.2. High-Throughput Sequencing
2.3. Sequence Analysis
3. Results
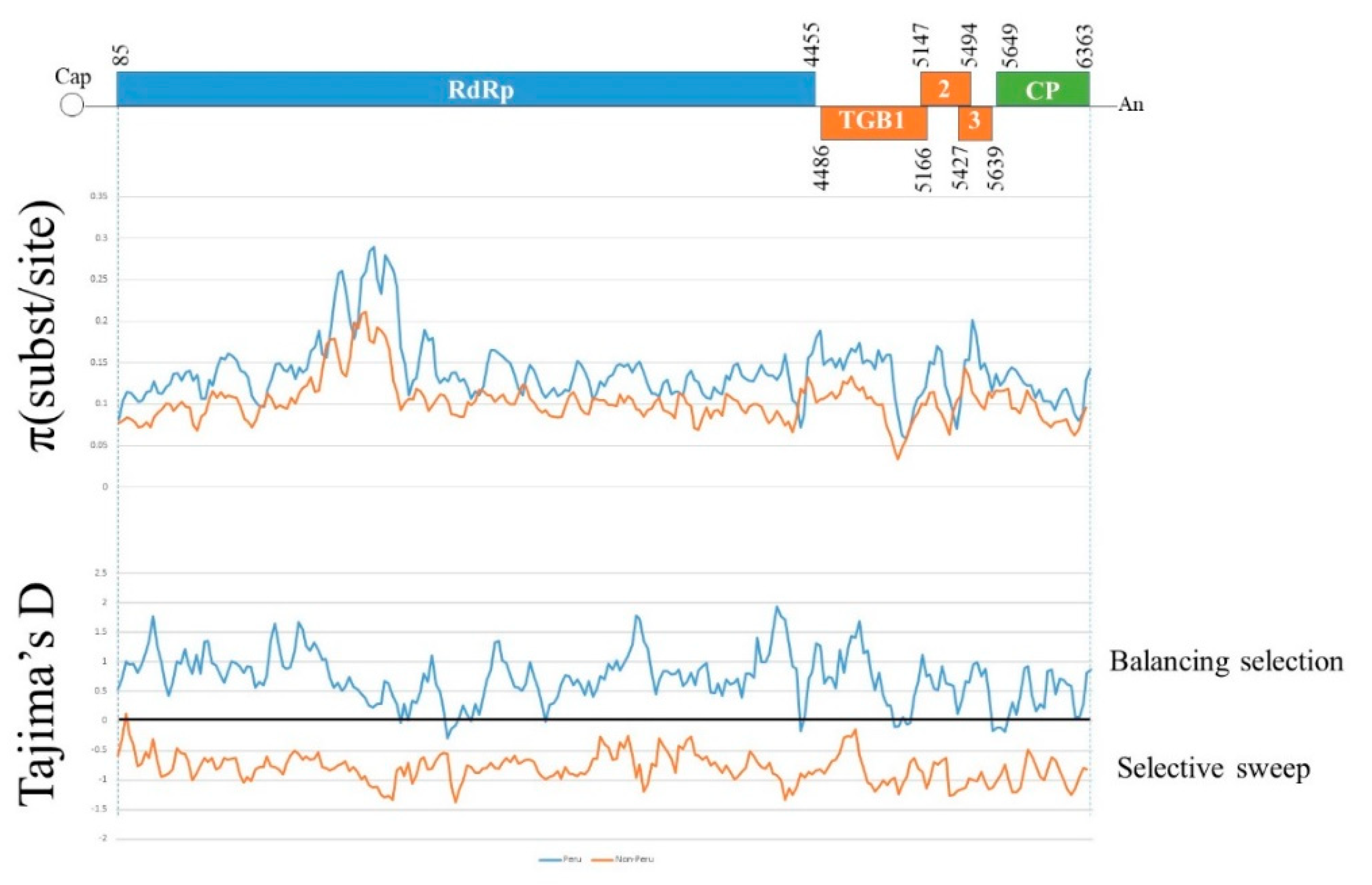
3.1. Sequence Alignments
3.2. Recombination Analyses
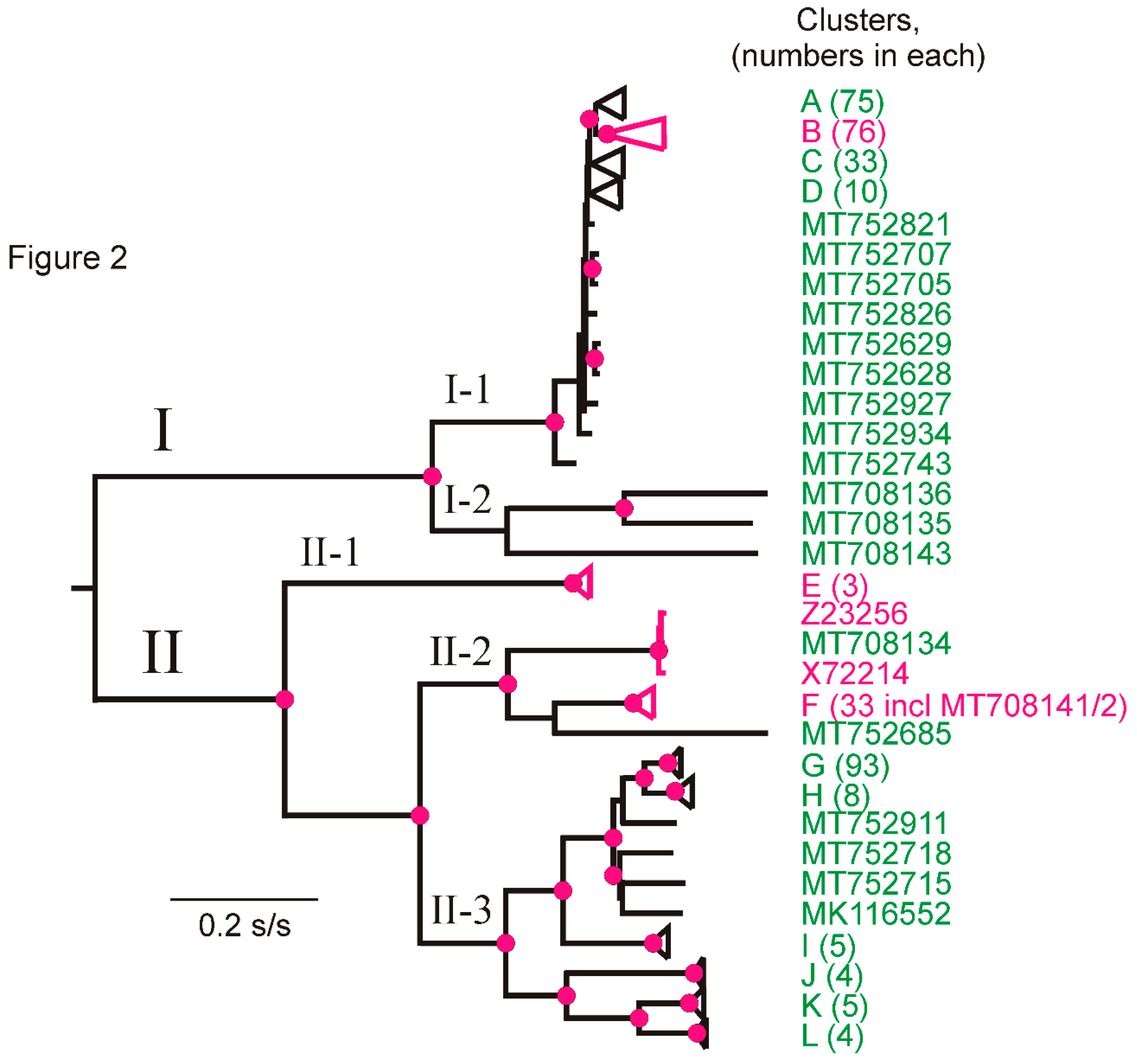
3.3. Phylogroups
3.4. World Populations of PVX
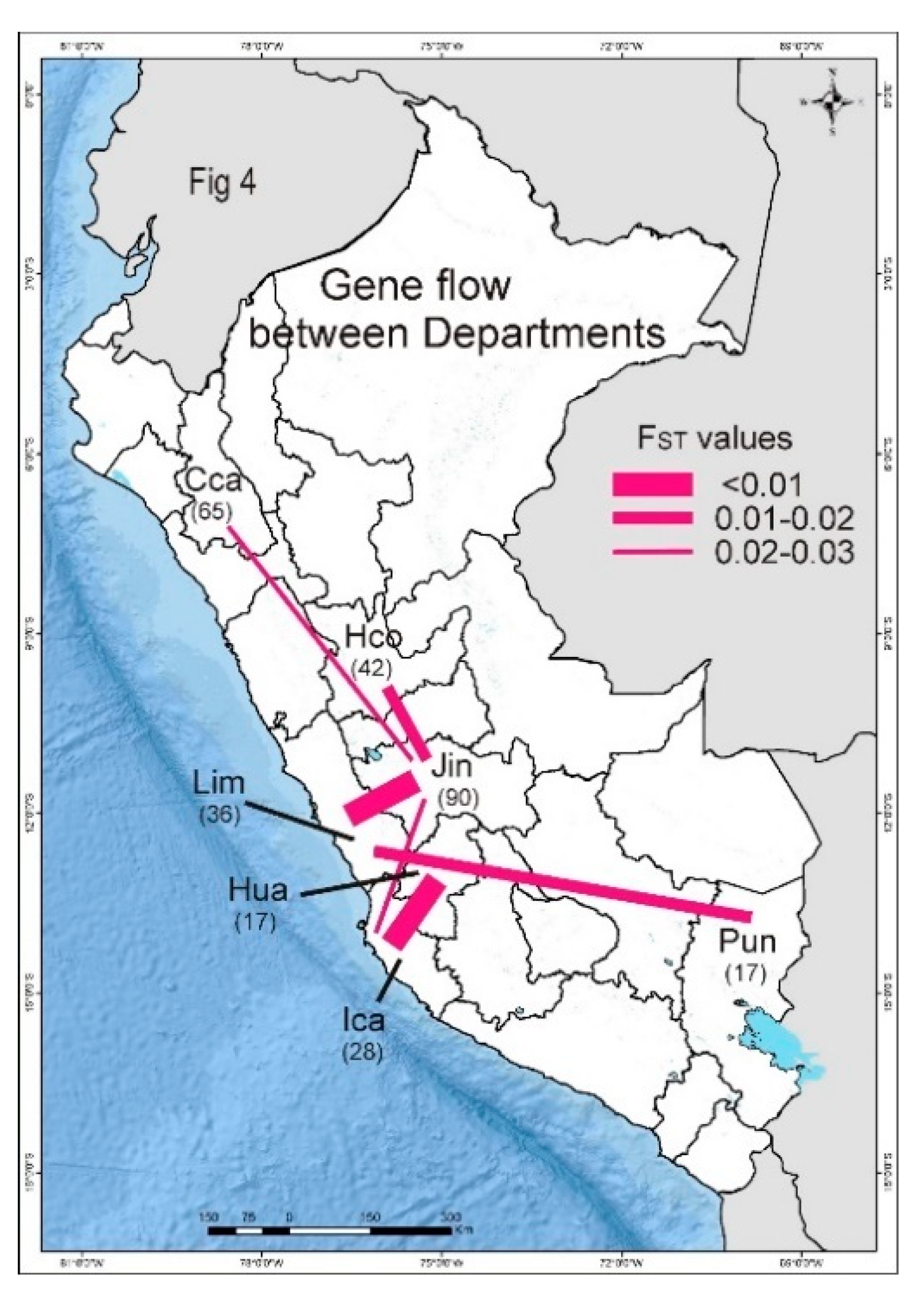
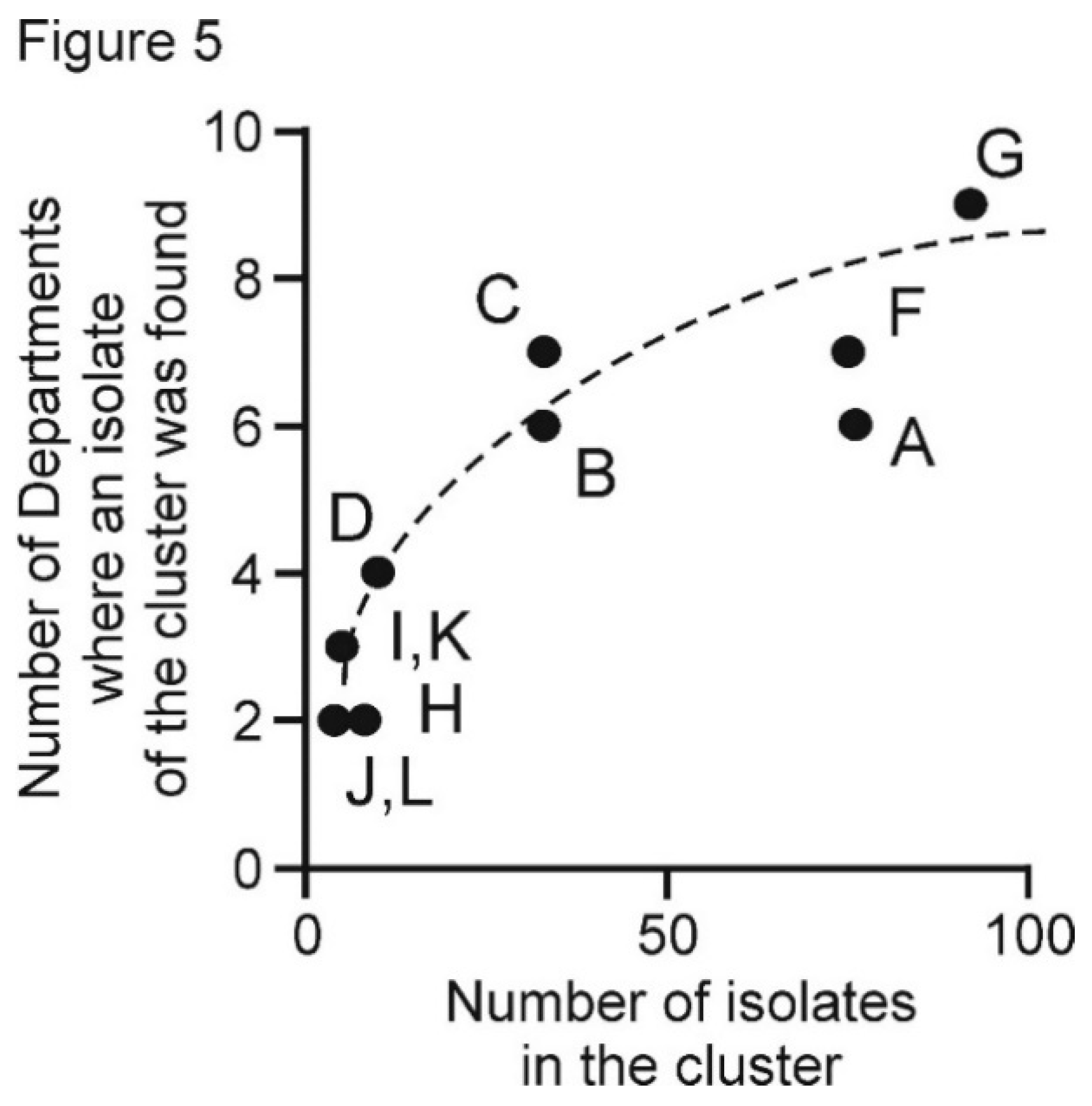
3.5. PVX Populations of Peru
3.6. Dating
3.7. Origin of PVX
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kreuze, J.F.; Vaira, A.M.; Menzel, W.; Candresse, T.; Zavriev, S.K.; Hammond, J.; Ryu, K.H.; Consortium, I.R. ICTV virus taxonomy profile: Alphaflexiviridae. J. Gen. Virol. 2020, 101, 699. [Google Scholar] [CrossRef]
- Martelli, G.P.; Adams, M.J.; Kreuze, J.F.; Dolja, V.V. Family Flexiviridae: A case study in virion and genome plasticity. Annu. Rev. Phytopathol. 2007, 45, 73–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salaman, R.N. The potato virus “X”: Its strains and reactions. Phil. Trans. Roy. Soc. Lond. Ser. B Biol. Sci. 1938, 229, 137–217. [Google Scholar]
- Salaman, R.N. Some notes on the history of curl. Tijdschr. Plantenziekten 1949, 55, 118–128. [Google Scholar] [CrossRef]
- Smith, K.M. On the composite nature of certain potato virus diseases of the mosaic group as revealed by the use of plant indicators and selective methods of transmission. Proc. Roy. Soc. Lond. Ser. B 1931, 109, 251–267. [Google Scholar]
- Smith, K.M. A Textbook of Plant Virus Diseases, 2nd ed.; Churchill Ltd.: London, UK, 1937. [Google Scholar]
- Bercks, R. Potato Virus X. CMI/AAB Descriptions of Plant Viruses No. 4; CABI: Wallingford, UK, 1970. [Google Scholar]
- De Bokx, J.A.; van der Want, J.P.H. (Eds.) Viruses of Potatoes and Seed Potato Production, 2nd ed.; Centre for Agricultural Publishing and Documentation: Wageningen, The Netherlands, 1987; 259p. [Google Scholar]
- Jones, R.A.C. Virus disease problems facing potato industries worldwide: Viruses found, climate change implications, rationalizing virus strain nomenclature, and addressing the potato virus Y issue. In The Potato: Botany, Production and Uses; Navarre, R., Pavek, M.J., Eds.; CABI: Wallingford, UK, 2014; pp. 202–224. [Google Scholar]
- Kreuze, J.; Souza-Dias, J.A.C.; Jeevalatha, A.; Figueira, A.R.; Valkonen, J.P.T.; Jones, R.A.C. Viral diseases in potato. In The Potato Crop; Campos, H., Ortiz, O., Eds.; Springer: Cham, Switzerland, 2020; pp. 389–430. [Google Scholar]
- Loebenstein, G.; Berger, P.H.; Brunt, A.A. (Eds.) Virus and Virus-Like Diseases of Potatoes and Production of Seed-Potatoes; Kluwer Academic Publishers: Dorercht, The Netherlands, 2001. [Google Scholar]
- Stevenson, W.R.; Loria, R.; Franc, G.D.; Weingartner, D.P. Compendium of Potato Diseases; American Phytopathological Society (APS) Press: St. Paul, MN, USA, 2001; pp. 72–77. [Google Scholar]
- Jones, R.A.C. Global plant virus disease pandemics and epidemics. Plants 2021, 10, 233. [Google Scholar] [CrossRef]
- Munro, J. Potato virus X. In Compendium of Potato Diseases; Hooker, W.J., Ed.; International Potato Center: Lima, Peru; American Phytopathological Society (APS) Press: St. Paul, MN, USA, 1981; pp. 72–77. [Google Scholar]
- Wilson, C.R.; Jones, R.A.C. Virus content of seed potato stocks produced in a unique seed potato production scheme. Ann. Appl. Biol. 1990, 116, 103–109. [Google Scholar] [CrossRef]
- Wilson, C.R.; Jones, R.A.C. Occurrence of potato virus X strain group 1 in seed stocks of potato cultivars lacking resistance genes. Ann. Appl. Biol. 1995, 127, 479–487. [Google Scholar] [CrossRef]
- Valkonen, J.P.T. Viruses: Economical losses and biotechnological potential. In Potato Biology and Biotechnology: Advances and Perspectives; Vreugdenhil, D., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2007; pp. 619–641. [Google Scholar]
- Wright, N.S. The effect of separate infections by potato viruses X and S on netted gem potato. Amer. Potato J. 1977, 54, 147–149. [Google Scholar] [CrossRef]
- Murphy, P.A.; McKay, R. The compound nature of crinkle and its production by means of a mixture of viruses. Sci. Proc. Roy. Dublin Soc. 1932, 5, 227–247. [Google Scholar]
- Rochow, W.; Ross, A.F. Virus multiplication in plants doubly infected by potato viruses X and Y. Virology 1955, 1, 10–27. [Google Scholar] [CrossRef]
- Nyalugwe, E.P.; Wilson, C.R.; Coutts, B.A.; Jones, R.A.C. Biological properties of Potato virus X in potato: Effects of mixed infection with potato virus S and resistance phenotypes in cultivars from three continents. Plant Dis. 2012, 96, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Kendall, A.; McDonald, M.; Bian, W.; Bowles, T.; Baumgarten, S.C.; Shi, J.; Stewart, P.L.; Bullitt, E.; Gore, D.; Irving, T.C. Structure of flexible filamentous plant viruses. J. Virol. 2008, 82, 9546–9554. [Google Scholar] [CrossRef] [Green Version]
- King, A.M.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier Science: Amsterdam, The Netherlands, 2011; Volume 9. [Google Scholar]
- Cockerham, G. Strains of potato virus X. In Proceedings of the Second Conference on Potato Virus Diseases, 1954; Lisse-Wageningen: Wageningen, The Netherlands, 1955; pp. 89–92. [Google Scholar]
- Mills, W. Inheritance of immunity to potato virus X. Am. Potato J. 1965, 42, 294–295. [Google Scholar]
- Solomon-Blackburn, R.M.; Barker, H. A review of host major-gene resistance to potato viruses X, Y, A and V in potato: Genes, genetics and mapped locations. Heredity 2001, 86, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Valkonen, J.P.T. Natural genes and mechanisms for resistance to viruses in cultivated and wild potato species (Solanum spp.). Plant Breed. 1994, 112, 1–16. [Google Scholar] [CrossRef]
- Adams, S.E.; Jones, R.A.C.; Coutts, R.H.A. Occurrence of resistance-breaking strains of potato virus X in potato stocks in England and Wales. Plant Pathol. 1984, 33, 435–437. [Google Scholar] [CrossRef]
- Davidson, T.M.W. Breeding for resistance to virus disease of the potato (Solanum tuberosum) at the Scottish Plant Breeding Station. Scott. Plant Breed. Stn. Annu. Rep. 1980, 100–108. [Google Scholar]
- Fribourg, C.E. Studies on potato virus X strains isolated from Peruvian potatoes. Potato Res. 1975, 18, 216–226. [Google Scholar] [CrossRef]
- Moreira, A.; Jones, R.A.C.; Fribourg, C.E. Properties of a resistance-breaking strain of potato virus X. Ann. Appl. Biol. 1980, 95, 93–103. [Google Scholar] [CrossRef]
- Jones, R.A.C. The ecology of viruses infecting wild and cultivated potatoes in the Andean region of South America. In Pests, Pathogens, and Vegetation; Thresh, J.M., Ed.; Pitman: London, UK, 1981; pp. 89–107. [Google Scholar]
- Adams, S.E.; Jones, R.A.C.; Coutts, R.H.A. Effect of temperature on potato virus X infection in potato cultivars carrying different combinations of hypersensitivity genes. Plant Pathol. 1986, 35, 517–526. [Google Scholar] [CrossRef]
- Tozzini, A.C.; Ceriani, M.; Cramer, P.; Palva, E.; Hopp, H.E. PVX MS, a new strain of potato virus that overcomes the extreme resistance gene Rx. J. Phytopathol. 1994, 141, 241–248. [Google Scholar] [CrossRef]
- Jones, R.A.C. Breakdown of potato virus X resistance gene Nx: Selection of a group four strain from strain group three. Plant Pathol. 1982, 31, 325–331. [Google Scholar] [CrossRef]
- Jones, R.A.C. Further studies on resistance-breaking strains of potato virus X. Plant Pathol. 1985, 34, 182–189. [Google Scholar] [CrossRef]
- Santa-Cruz, S.; Baulcombe, D.C. Molecular analysis of potato virus X isolates in relation to the potato hypersensitivity gene Nx. Mol. Plant Microbe Interact. 1993, 6, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Goulden, M.G.; Köhm, B.A.; Santa-Cruz, S.; Kavanagh, T.A.; Baulcombe, D.C. A feature of the coat protein of potato virus X affects both induced virus resistance in potato and viral fitness. Virology 1993, 197, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, T.A.; Goulden, M.G.; Santa Cruz, S.; Chapman, S.; Barker, I.; Baulcombe, D.C. Molecular analysis of a resistance-breaking strain of potato virus X. Virology 1992, 189, 609–617. [Google Scholar] [CrossRef]
- Malcuit, I.; Marano, M.R.; Kavanagh, T.A.; De Jong, W.; Forsyth, A.; Baulcombe, D.C. The 25-kDa movement protein of PVX elicits Nb-mediated hypersensitive cell death in potato. Mol. Plant Microbe Interact. 1999, 12, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Querci, M.; Baulcombe, D.C.; Goldbach, R.W.; Salazar, L.F. Analysis of the resistance-breaking determinants of potato virus X (PVX) strain HB on different potato genotypes expressing extreme resistance to PVX. Phytopathology 1995, 85, 1003–1010. [Google Scholar] [CrossRef]
- Cox, B.A.; Jones, R.A.C. Genetic variability in the coat protein gene of Potato virus X and the current relationship between phylogenetic placement and resistance groupings. Arch. Virol. 2010, 155, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Kutnjak, D.; Silvestre, R.; Cuellar, W.; Perez, W.; Müller, G.; Ravnikar, M.; Kreuze, J. Complete genome sequences of new divergent potato virus X isolates and discrimination between strains in a mixed infection using small RNAs sequencing approach. Virus Res. 2014, 191, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, D.; Gutierrez-Sanchez, P.; Marin-Montoya, M. Phylogenetic analysis and molecular variability of potato virus X (PVX) in potato crops of Antioquia. Acta Biol. Colomb. 2016, 21, 111–122. [Google Scholar]
- Gutiérrez, P.A.; Alzate, J.F.; Montoya, M.M. Complete genome sequence of an isolate of Potato virus X (PVX) infecting Cape gooseberry (Physalis peruviana) in Colombia. Virus Genes 2015, 50, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, J.G. The Potato: Evolution, Biodiversity and Genetic Resources; Belhaven Press: London, UK, 1990. [Google Scholar]
- Nunn, N.; Qian, N. The Columbian exchange: A history of disease, food, and ideas. J. Econ. Perspect. 2010, 24, 163–188. [Google Scholar] [CrossRef] [Green Version]
- Glenndinning, D.R. Potato Introductions and Breeding up to the 20th Century. New Phytol. 1983, 94, 479–505. [Google Scholar] [CrossRef]
- Kahn, R.P.; Hewitt, W.B.; Goheen, A.C.; Wallace, J.M.; Roistacher, C.N.; Neuer, E.M.; Brierley, P.; Cochran, L.C.; Monroe, R.L.; Ackermann, W.L.; et al. Detection of viruses in foreign plant introductions under quarantine in the United States. Plant Dis. Reptr. 1963, 47, 261–265. [Google Scholar]
- McKee, R.K. Virus infection in South American potatoes. Eur. Potato J. 1964, 7, 145–151. [Google Scholar] [CrossRef]
- Monasterios de la Torre, T. Presence of viruses in Bolivian potatoes. Turrialba 1966, 16, 257–260. [Google Scholar]
- Silberschmidt, K. The spontaneous occurence of strains of potato virus X and Y in South America. J. Phytopathol. 1961, 42, 175–192. [Google Scholar]
- Kahn, R.P.; Monroe, R.; Hewitt, W.B.; Goheen, A.C.; Wallace, J.M.; Roistacher, C.N.; Neuer, E.M.; Ackerman, W.L.; Winters, H.F.; Seaton, C.A.; et al. Incidence of virus detection in vegetatively propagated plant introductions under quarantine in the United States, 1957–1967. Plant Dis. Rep. 1967, 51, 715. [Google Scholar]
- Kahn, R.P.; Monroe, R.L. Virus infection in plant introductions collected as vegetative propagations: I. Wild vs. cultivated Solanum species. Plant Prot. Bull. FAO 1970, 18, 97–101. [Google Scholar]
- Bertschinger, L.; Scheidegger, U.C.; Luther, K.; Pinillos, O.; Hidalgo, A. Virus incidence in native and modern cultivars in the Peruvian highlands. Rev. Latinoam. Papa 1990, 3, 62–79. [Google Scholar] [CrossRef]
- Cockerham, G. Genetical studies on resistance to potato viruses X and Y. Heredity 1970, 25, 309–348. [Google Scholar] [CrossRef]
- Valkonen, J.P.T.; Jones, R.A.C.; Slack, S.A.; Watanabe, K.N. Resistance specificities to viruses in potato: Standardization of nomenclature. Plant Breed. 1996, 115, 433–438. [Google Scholar] [CrossRef]
- Fuentes, S.; Gibbs, A.J.; Adams, I.P.; Wilson, C.; Botermans, M.; Fox, A.; Kreuze, J.; Boonham, N.; Kehoe, M.A.; Jones, R.A.C. Potato virus A isolates from three continents: Their biological properties, phylogenetics, and prehistory. Phytopathology 2021, 111, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, S.; Jones, R.A.C.; Matsuoka, H.; Ohshima, K.; Kreuze, J.; Gibbs, A.J. Potato virus Y; the Andean connection. Virus Evol. 2019, 5, vez037. [Google Scholar] [CrossRef]
- Santillan, F.W.; Fribourg, C.E.; Adams, I.P.; Gibbs, A.J.; Boonham, N.; Kehoe, M.A.; Maina, S.; Jones, R.A.C. The biology and phylogenetics of Potato virus S isolates from the Andean region of South America. Plant Dis. 2018, 102, 869–885. [Google Scholar] [CrossRef] [Green Version]
- Fox, A.; Fowkes, A.R.; Skelton, A.; Harju, V.; Buxton-Kirk, A.; Kelly, M.; Forde, S.M.D.; Pufal, H.; Conyers, C.; Ward, R.; et al. Using high-throughput sequencing in support of a plant health outbreak reveals novel viruses in Ullucus tuberosus (Basellaceae). Plant Pathol. 2019, 68, 576–587. [Google Scholar]
- Chen, Y.-R.; Zheng, Y.; Liu, B.; Zhong, S.; Giovannoni, J.; Fei, Z. A cost-effective method for Illumina small RNA-Seq library preparation using T4 RNA ligase 1 adenylated adapters. Plant Methods 2012, 8, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.-S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Abascal, F.; Zardoya, R.; Telford, M.J. Translator X: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38 (Suppl. 2), W7–W13. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Holmes, E.C.; Worobey, M.; Rambaut, A. Phylogenetic evidence for recombination in dengue virus. Mol. Biol. Evol. 1999, 16, 405–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Lott, M.; Martin, D.P.; Moulton, V. Identifying recombinants in human and primate immunodeficiency virus sequence alignments using quartet scanning. BMC Bioinform. 2009, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retr. 2005, 21, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- McGuire, G.; Wright, F. TOPAL 2.0: Improved detection of mosaic sequences within multiple alignments. Bioinformatics 2000, 16, 130–134. [Google Scholar] [CrossRef] [Green Version]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Jeanmougin, F.; Thompson, J.D.; Gouy, M.; Higgins, D.G.; Gibson, T.J. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 1998, 23, 403–405. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114. [Google Scholar] [CrossRef] [Green Version]
- Fourment, M.; Gibbs, M.J. PATRISTIC: A program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evol. Biol. 2006, 6, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Rozas, J. DNA sequence polymorphism analysis using DnaSP. In Bioinformatics for DNA Sequence Analysis; Posada, D., Ed.; Springer: Cham, Switzerland, 2009; pp. 337–350. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.R.; Slatkin, M.; Maddison, W.P. Estimation of levels of gene flow from DNA sequence data. Genetics 1992, 132, 583–589. [Google Scholar] [CrossRef]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- To, T.-H.; Jung, M.; Lycett, S.; Gascuel, O. Fast dating using least-squares criteria and algorithms. Syst. Biol. 2016, 65, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Karpova, O.V.; Arkhipenko, M.V.; Zayakina, O.V.; Nikitin, N.A.; Kiselyova, O.I.; Kozlovsky, S.V.; Rodionova, N.P.; Atabekov, J.G. Regulation of RNA translation in potato virus X RNA-coat protein complexes: The key role of the N-terminal segment of the protein. Mol. Biol. 2006, 40, 628–634. [Google Scholar] [CrossRef]
- Chapman, S.; Kavanagh, T.; Baulcombe, D.C. Potato virus X as a vector for gene expression in plants. Plant J. 1992, 2, 549–557. [Google Scholar]
- Santa-Cruz, S.; Roberts, A.G.; Prior, D.A.; Chapman, S.; Oparka, K.J. Cell-to-cell and phloem-mediated transport of potato virus X: The role of virions. Plant Cell 1998, 10, 495–510. [Google Scholar] [CrossRef] [Green Version]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics; Sinauer Associates: Sunderland, MA, USA, 1997; Volume 116, 635p. [Google Scholar]
- Wright, S. Evolution and the Genetics of Populations, Volume 4: Variability Within and Among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1984. [Google Scholar]
- Herrerías-Azcué, F.; Pérez-Muñuzuri, V.; Galla, T. Stirring does not make populations well mixed. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fribourg, C.E.; Gibbs, A.J.; Adams, I.P.; Boonham, N.; Jones, R.A.C. Biological and molecular properties of wild potato mosaic virus isolates from pepino (Solanum muricatum). Plant Dis. 2019, 103, 1746–1756. [Google Scholar] [CrossRef]
- Hajizadeh, M.; Sokhandan-Bashir, N. Population genetic analysis of potato virus X based on the CP gene sequence. Virus Disease 2017, 28, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, J.S. Great Irish Potato Famine; The History Press: Dublin, Ireland, 2002. [Google Scholar]
- Zadoks, J.C. The potato murrain on the European continent and the revolutions of 1848. Potato Res. 2008, 51, 5–45. [Google Scholar] [CrossRef]
- Gutaker, R.M.; Weiß, C.L.; Ellis, D.; Anglin, N.L.; Knapp, S.; Fernández-Alonso, J.L.; Prat, S.; Burbano, H.A. The origins and adaptation of European potatoes reconstructed from historical genomes. Nat. Ecol. Evol. 2019, 3, 1093–1101. [Google Scholar] [CrossRef]
- Spooner, D.M.; Núñez, J.; Trujillo, G.; del Rosario Herrera, M.; Guzmán, F.; Ghislain, M. Extensive simple sequence repeat genotyping of potato landraces supports a major reevaluation of their gene pool structure and classification. Proc. Natl. Acad. Sci. USA 2007, 104, 19398–19403. [Google Scholar] [CrossRef] [Green Version]
- Ovchinnikova, A.; Krylova, E.; Gavrilenko, T.; Smekalova, T.; Zhuk, M.; Knapp, S.; Spooner, D.M. Taxonomy of cultivated potatoes (Solanum section Petota: Solanaceae). Bot. J. Linn. Soc. 2011, 165, 107–155. [Google Scholar] [CrossRef] [Green Version]
- Hardigan, M.A.; Laimbeer, F.P.E.; Newton, L.; Crisovan, E.; Hamilton, J.P.; Vaillancourt, B.; Wiegert-Rininger, K.; Wood, J.C.; Douches, D.S.; Farré, E.M. Genome diversity of tuber-bearing Solanum uncovers complex evolutionary history and targets of domestication in the cultivated potato. Proc. Natl. Acad. Sci. USA 2017, 114, E9999–E10008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browman, D.L. Toward the development of the Tiahuanaco (Tiwanaku) state. In Advances in Andean Archeology; Browman, D.L., Ed.; De Gruyter Mouton: Berlin, Germany, 1978; pp. 327–349. [Google Scholar]
- Hawkes, J.G. History of the potato. In The Potato Crop: The Scientific Basis for Improvement; Harris, P.M., Ed.; Springer: Dordrecht, The Netherlands, 1978; pp. 1–14. [Google Scholar]
- Ohshima, K.; Tomitaka, Y.; Wood, J.T.; Minematsu, Y.; Kajiyama, H.; Tomimura, K.; Gibbs, A.J. Patterns of recombination in turnip mosaic virus genomic sequences indicate hotspots of recombination. J. Gen. Virol. 2007, 88, 298–315. [Google Scholar] [CrossRef]
- Santa-Cruz, S.; Baulcombe, D.C. Analysis of potato virus X coat protein genes in relation to resistance conferred by the genes Nx, Nb and Rx1 of potato. J. Gen. Virol. 1995, 76, 2057–2061. [Google Scholar] [CrossRef]
- Jones, R.A.C.; Boonham, N.; Adams, I.P.; Fox, A. Historical virus isolate collections: An invaluable resource connecting plant virology’s pre-sequencing and post-sequencing eras. Plant Pathol. 2021, 70, 235–248. [Google Scholar] [CrossRef]
- Kehoe, M.A.; Jones, R.A.C. Improving Potato virus Y strain nomenclature: Lessons from comparing isolates obtained over a 73-year period. Plant Pathol. 2016, 65, 322–333. [Google Scholar] [CrossRef]
- Gibbs, A.J.; Ohshima, K.; Yasaka, R.; Mohammadi, M.; Gibbs, M.J.; Jones, R.A.C. The phylogenetics of the global population of potato virus Y and its necrogenic recombinants. Virus Evol. 2017, 3, vex002. [Google Scholar] [CrossRef] [Green Version]
- Green, K.; Quintero-Ferrer, A.; Chikh-Ali, M.; Jones, R.A.C.; Karasev, A.V. Genetic diversity of nine non-recombinant potato virus Y Isolates from three biological strain groups: Historical and geographical insights. Plant Dis. 2020, 104, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Cockerham, G. The reactions of potato varieties to viruses X, A, B and C. Ann. Appl. Biol. 1943, 30, 338–344. [Google Scholar] [CrossRef]
- Bawden, F.C. The viruses causing top necrosis (acronecrosis) of the potato. Ann. Appl. Biol. 1936, 23, 487–497. [Google Scholar] [CrossRef]
- Bawden, F.C.; Sheffield, F.M.L. The relationships of some viruses causing necrotic diseases of the potato. Ann. Appl. Biol. 1944, 31, 33–40. [Google Scholar] [CrossRef]
- Harrison, B.D. Plant virus ecology: Ingredients, interactions and environmental influences. Ann. Appl. Biol. 1981, 99, 195–209. [Google Scholar] [CrossRef]
- De Cubillos, C.F.; Thurston, H. The effect of viruses on infection by Phytophthora infestans (Mont.) de Bary in potatoes. Am. Potato J. 1975, 52, 221–226. [Google Scholar] [CrossRef]
- Nie, X.; Singh, R.P. Detection of multiple potato viruses using an oligo (dT) as a common cDNA primer in multiplex RT-PCR. J. Virol. Methods 2000, 86, 179–185. [Google Scholar] [CrossRef]
- Mortimer-Jones, S.M.; Jones, M.G.K.; Jones, R.A.C.; Thomson, G.; Dwyer, G.I. A single tube, quantitative real-time RT-PCR assay that detects four potato viruses simultaneously. J. Virol. Methods 2009, 161, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Maina, S.; Zheng, L.; Rhodoni, B.C. Targeted genome sequencing (TG-Seq) approaches to detct plant viruses. Viruses 2021, 13, 593. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Isolate | Source Species | Cultivar/Breeding Line | Accession Number | Where Collected/Obtained | Isolation Year | Strain Group (=Pathotype) | GenBank Code | References |
| E | S. tuberosum subsp. andigena | Renacimiento | N/A | Central- southern highlands- Perú | 1973 | N/A | MT708135 | [30] |
| CP (=C) | S. tuberosum subsp. andigena | Renacimiento | N/A | Central- southern highlands-Perú | 1973 | 2 | MT708142 | [30] |
| CP4 | S. tuberosum subsp. andigena | (Renacimiento)** | N/A | (Central- southern highlands-Perú) | (1973) | 4 | MT708141 | [36] |
| DP (=D) | S. goniocalyx | Runtush | 0CH 02736 | Jauja, Junin, Department, Perú | 1973 | 1 and 3 (mixture) | MT708143 | [30] |
| A | S. tuberosum subsp. andigena | Ccompis | PI 308884 | Wisconsin, USA in tuber from Peru * | 1970 | 1 and 3(mixture) | MT708136 | [30] |
| HB | S. tuberosum subsp. andigena | Suta | N/A | Puna, Potosi Department, Bolivia | 1975 | 4 | MT708134 | [31] |
| B | S. tuberosum subsp. tuberosum | Duke of York | N/A | Scotland | 1940 | 2 | MT708140 | [24,36] |
| DX | S. tuberosum subsp. tuberosum | Desiree | N/A | Cambridgeshire, England | 1980 | 3 | No sequence | [35] |
| DX4 | S. tuberosum subsp. tuberosum | (Desiree) | N/A | (Cambridgeshire, England) | (1980) | 4 | MT708139 | [35] |
| EX | S. tuberosum subsp. tuberosum | Epicure | N/A | Cambridgeshire, England | 1983 | 2 | MT708138 | [28,36] |
| EX4 | S. tuberosum subsp. tuberosum | (Epicure) | N/A | (Cambridgeshire, England) | (1983) | 4 | MT708137 | [36] |
| (b) | ||||||||
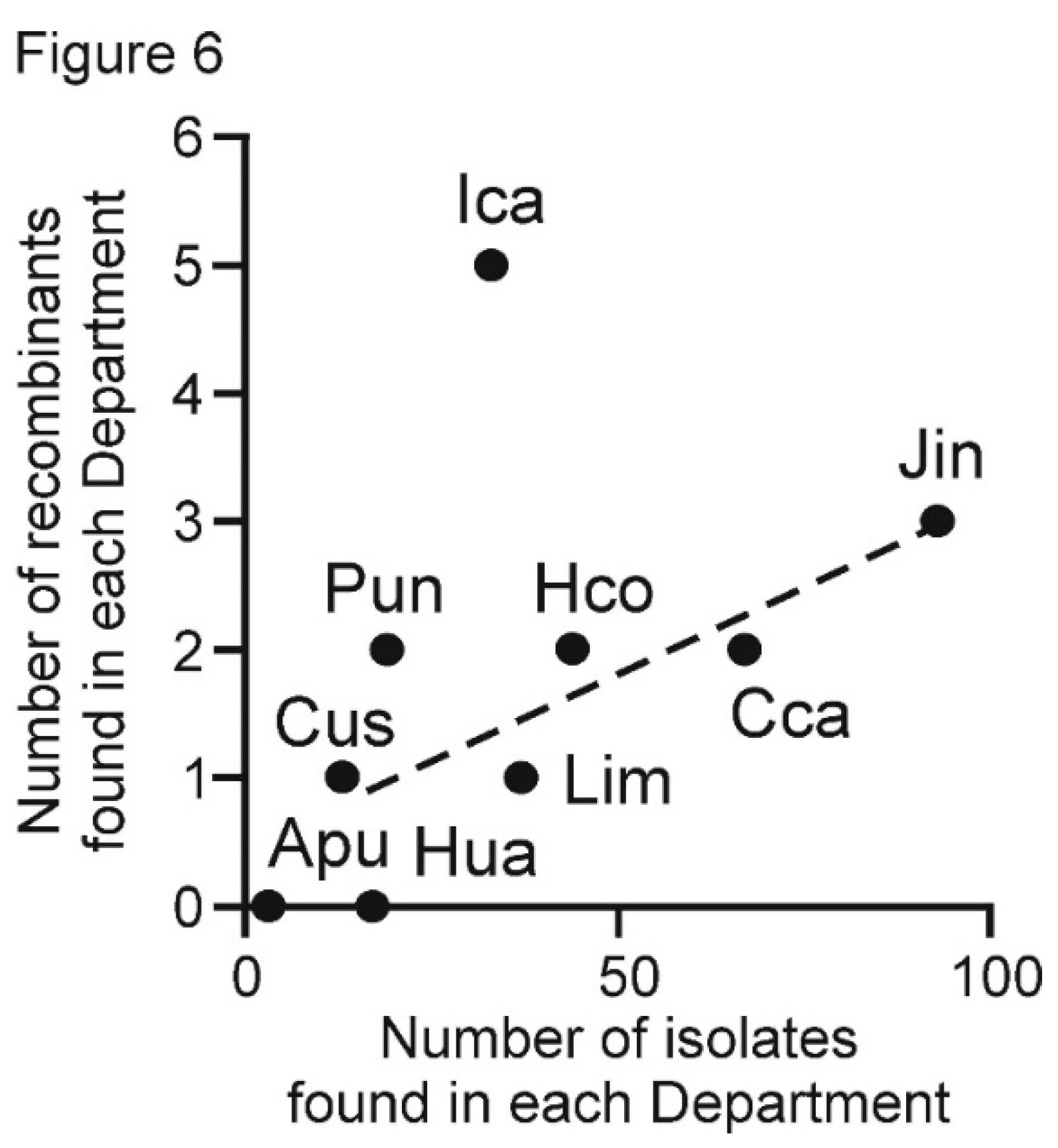
| Isolate Prefix | Peruvian Department Collected From | Year of Isolation | Total Samples | Number of Isolates Sequenced | ||||
| Apu | Apurimac | 2019 | 3 | 3 | ||||
| Cca | Cajamarca | 2016 | 60 | 67 | ||||
| Cus | Cusco | 2016 | 10 | 13 | ||||
| Hua | Huancavelica | 2016–2018 | 15 | 17 | ||||
| Hco | Huanuco | 2016 | 37 | 44 | ||||
| Ica | Ica | 2017 | 26 | 33 | ||||
| Jin | Junin | 2016 | 77 | 93 | ||||
| Lim | Lima | 2017 | 29 | 37 | ||||
| Pun | Puno | 2018 | 12 | 19 | ||||
| TOTAL | 269 | 326 | ||||||
| Recombinant (Rec) | Major Parent | Minor Parent | Rec. Region | RDP4 Programs 1 | Method | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession (Acc.) Code | Isolate | Collection Site 2 | Acc. Code | Cluster | Isolate | Collection Site 2 | Acc. Code | Cluster | Isolate | Collection Site 2 | start | end | CRS 3 | |
| MT752615 | Cca004-2 | 5 | MT752839 | B | Jin125 | 229 | MT752614 | G | Cca004-1 | 4 | 2602 | 2812 | 7 | 0.739 |
| MT752631 | Cca043 | 21 | MT752857 | rec | Jin163 | 247 | MT752799 | K | Jin051 | 189 | 3530 | 3664 | 6 | 0.672 |
| MT752689 | Cus089-2 | 79 | MT752857 | rec | Jin163 | 247 | MT752757 | K | Ica016 | 147 | 3458 | 3562 | 4 | 0.667 |
| MT752729 | Hco027-1 | 119 | MT752785 | F | Ica099-1 | 175 | MT752872 | B | Jin174-3 | 262 | 5634 | 6387 | 7 | 0.719 |
| MT752730 | Hco027-2 | 120 | MT752873 | B | Jin175 | 263 | MT752785 | F | Ica099-1 | 175 | 5634 | 6387 | 7 | 0.719 |
| MT752758 | Ica017-1 | 148 | MT752611 | G | Apu008 | 1 | MT752791 | A | Jin035 | 181 | 5078 | 5237 | 5 | 0.693 |
| MT752761 | Ica027-1 | 151 | MT752790 | L | Jin032 | 180 | MT752763 | rec | Ica027-3 | 153 | 6117 | 6210 | 6 | 0.742 |
| MT752762 | Ica027-2 | 152 | MT752787 | A | Ica100 | 177 | MT752790 | L | Jin032 | 180 | 2918 | 3095 | 5 | 0.59 |
| MT752763 | Ica027-3 | 153 | MT752762 | rec | Ica027-2 | 152 | MT752761 | rec | Ica027-1 | 151 | 3513 | 3661 | 7 | 0.581 |
| MT752783 | Ica098-1 | 173 | MT752792 | G | Jin041 | 182 | AB196001 | B | Japan | - | 1580 | 1649 | 5 | 0.697 |
| MT752857 | Jin163 | 247 | MT752846 | C | Jin170B | 236 | MT752799 | K | Jin051 | 189 | 6258 | 6387 | 6 | 0.66 |
| MT752869 | Jin173 | 259 | MT752824 | C | Jin109-2 | 214 | MT752825 | I | Jin110 | 215 | 2346 | 2502 | 6 | 0.737 |
| MT752877 | Jin178 | 267 | MT752826 | S | Jin110-B | 216 | MT752825 | I | Jin110 | 215 | 2346 | 2601 | 7 | 0.738 |
| MT752896 | Lim084 | 286 | MT752804 | F | Jin059 | 194 | MT752829 | I | Jin113 | 219 | 5014 | 5521 | 7 | 0.696 |
| MT752919 | Pun001-2 | 309 | MT752921 | C | Pun002-2 | 311 | MT708136 | S | Peru | -4 | 5914 | 6073 | 6 | 0.644 |
| MT752933 | Pun035-2 | 323 | MT752934 | S | Pun035-3 | 324 | MT752774 | G | Ica040A | 164 | 1542 | 1652 | 6 | 0.672 |
| HQ450387 | USA | - | unknown | - | unknown | - | M95516 | B | UK | - | 1436 | 3731 | 6 | 0.573 |
| M31541 | Peru | - | X55802 | F | Argentina | - | unknown | - | - | 5387 | 5929 | 4 | 0.701 | |
| (a) | |||||||
| Continent | n | FST | Nm | ||||
| Asia | Europe | Andean South America | Asia | Europe | Andean South America | ||
| Africa | 6 | 0.079 | 0.201 | 0.318 | 2.88 | 0.99 | 0.54 |
| Asia | 15 | 0.189 | 0.308 | 1.07 | 0.56 | ||
| Europe | 17 | 0.073 | 3.19 | ||||
| Andean South America | 346 | ||||||
| (b) | |||||||
| Continent | n | FST | |||||
| East Asia | West Eurasia | Indian Subcontinent | Andean Region | ||||
| East Asia | 37 | 0.041 | 0.113 | 0.316 | |||
| West Eurasia | 53 | 0.069 | 0.205 | ||||
| Indian Subcontinent | 66 | 0.320 | |||||
| Andean Region | 313 | ||||||
| Department a | n | FST | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Cca | Cus | Hco | Hua | Ica | Jin | Lim | Pun | ||
| Apu | 3 | 0.024 | −0.224 | 0.045 | −0.237 | −0.137 | −0.062 | −0.109 | −0.017 |
| Cca | 65 | 0.191 | −0.008 | 0.183 | 0.116 | 0.021 | 0.075 | −0.005 | |
| Cus | 12 | 0.198 | −0.048 | −0.002 | 0.086 | 0.032 | 0.134 | ||
| Hco | 42 | 0.192 | 0.108 | 0.016 | 0.068 | −0.016 | |||
| Hua | 17 | 0.004 | 0.086 | 0.031 | 0.126 | ||||
| Ica | 28 | 0.027 | −0.009 | 0.049 | |||||
| Jin | 90 | 0.009 | −0.009 | ||||||
| Lim | 36 | 0.0.12 | |||||||
| Pun | 17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuentes, S.; Gibbs, A.J.; Hajizadeh, M.; Perez, A.; Adams, I.P.; Fribourg, C.E.; Kreuze, J.; Fox, A.; Boonham, N.; Jones, R.A.C. The Phylogeography of Potato Virus X Shows the Fingerprints of Its Human Vector. Viruses 2021, 13, 644. https://doi.org/10.3390/v13040644
Fuentes S, Gibbs AJ, Hajizadeh M, Perez A, Adams IP, Fribourg CE, Kreuze J, Fox A, Boonham N, Jones RAC. The Phylogeography of Potato Virus X Shows the Fingerprints of Its Human Vector. Viruses. 2021; 13(4):644. https://doi.org/10.3390/v13040644
Chicago/Turabian StyleFuentes, Segundo, Adrian J. Gibbs, Mohammad Hajizadeh, Ana Perez, Ian P. Adams, Cesar E. Fribourg, Jan Kreuze, Adrian Fox, Neil Boonham, and Roger A. C. Jones. 2021. "The Phylogeography of Potato Virus X Shows the Fingerprints of Its Human Vector" Viruses 13, no. 4: 644. https://doi.org/10.3390/v13040644
APA StyleFuentes, S., Gibbs, A. J., Hajizadeh, M., Perez, A., Adams, I. P., Fribourg, C. E., Kreuze, J., Fox, A., Boonham, N., & Jones, R. A. C. (2021). The Phylogeography of Potato Virus X Shows the Fingerprints of Its Human Vector. Viruses, 13(4), 644. https://doi.org/10.3390/v13040644