
Genomic Surveillance of Circulating SARS-CoV-2 in South East Italy: A One-Year Retrospective Genetic Study

 , ,
, ,  , ,
, ,  and
and 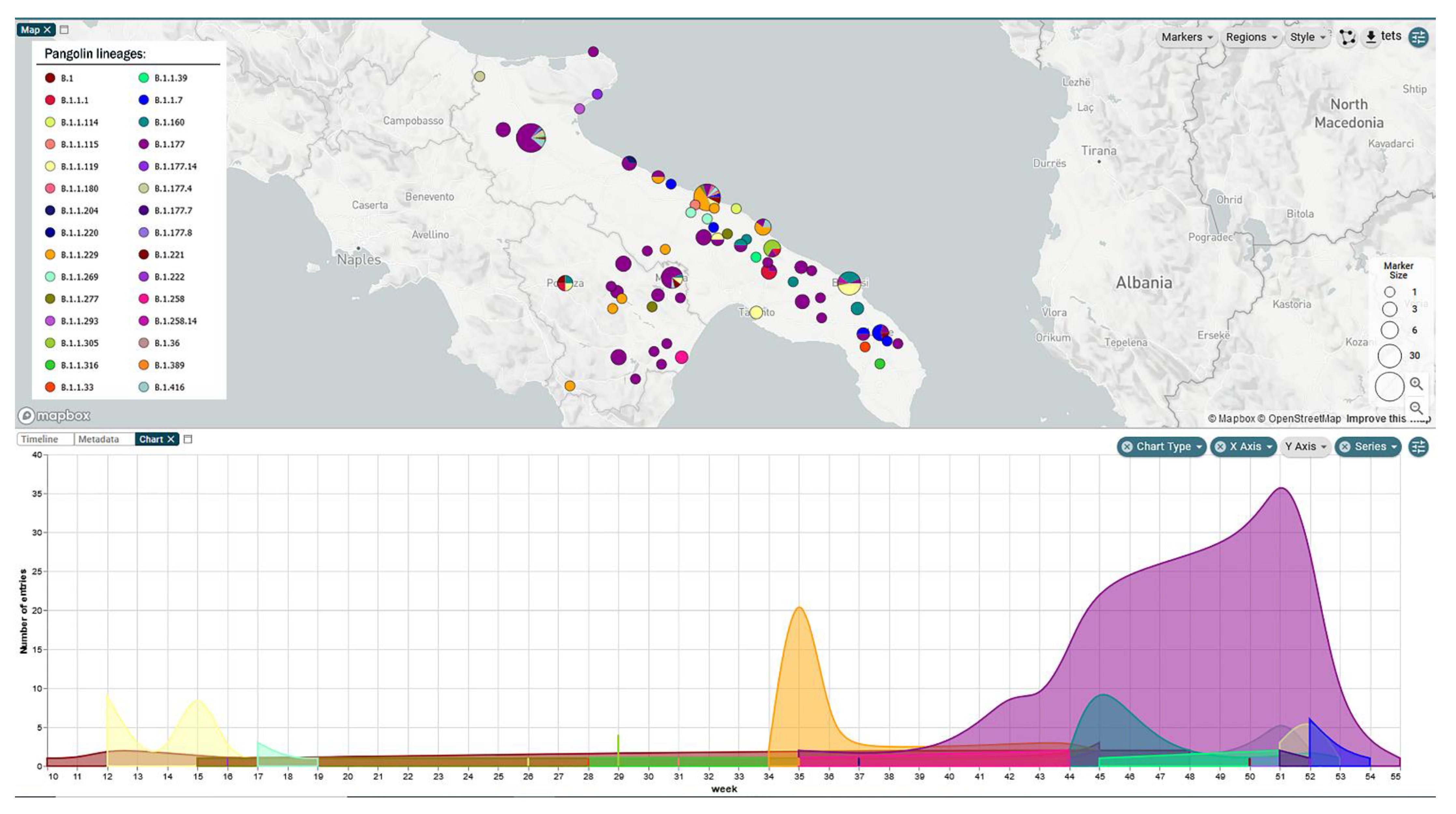{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collection and Testing
2.2. cDNA Synthesis and Viral Genome Amplification
2.3. Library Preparation and Whole-Genome Sequencing
2.4. Sequence Data Analysis
2.5. Phylogenetic Analysis
2.6. Ethical Statement
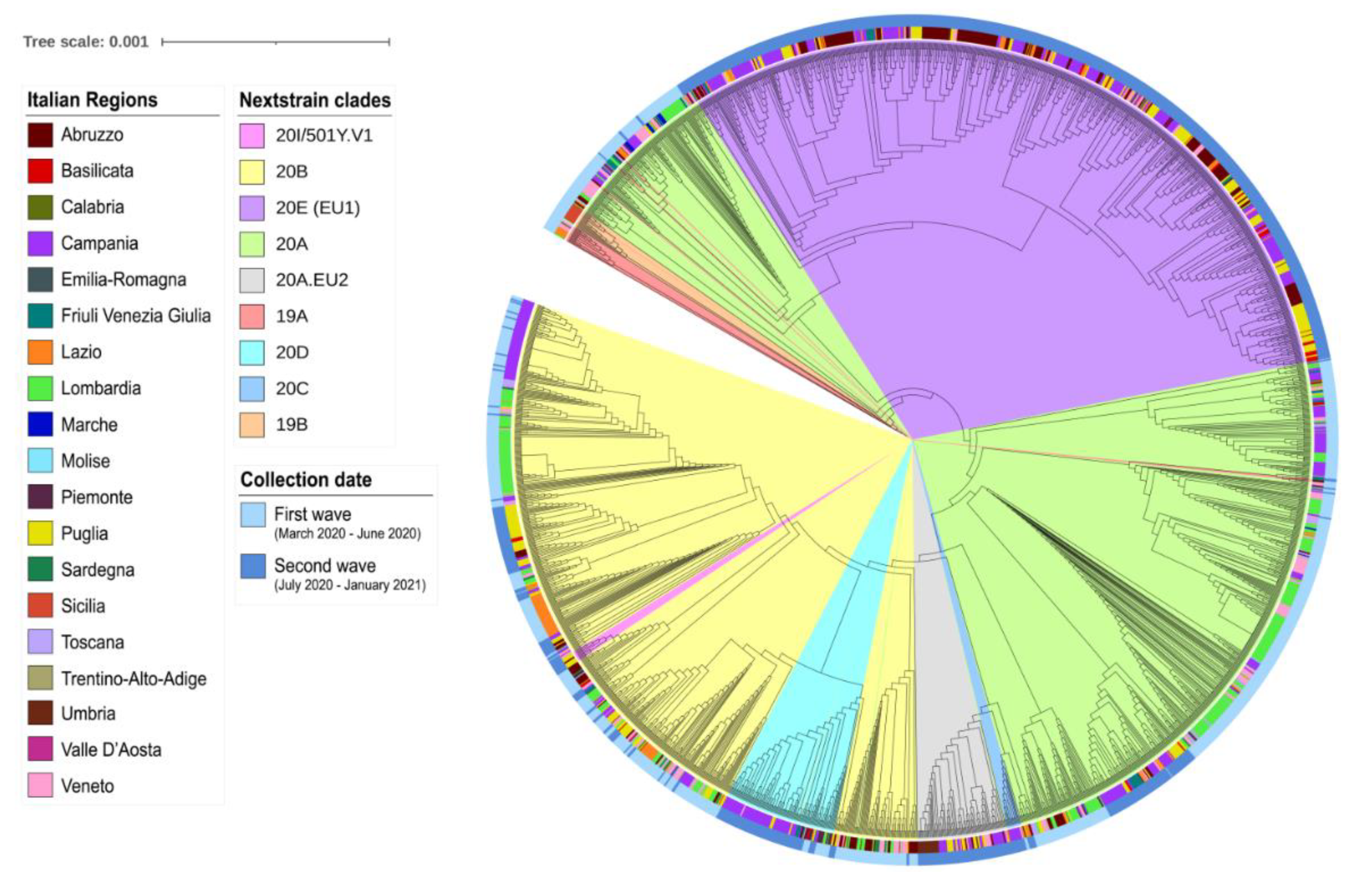
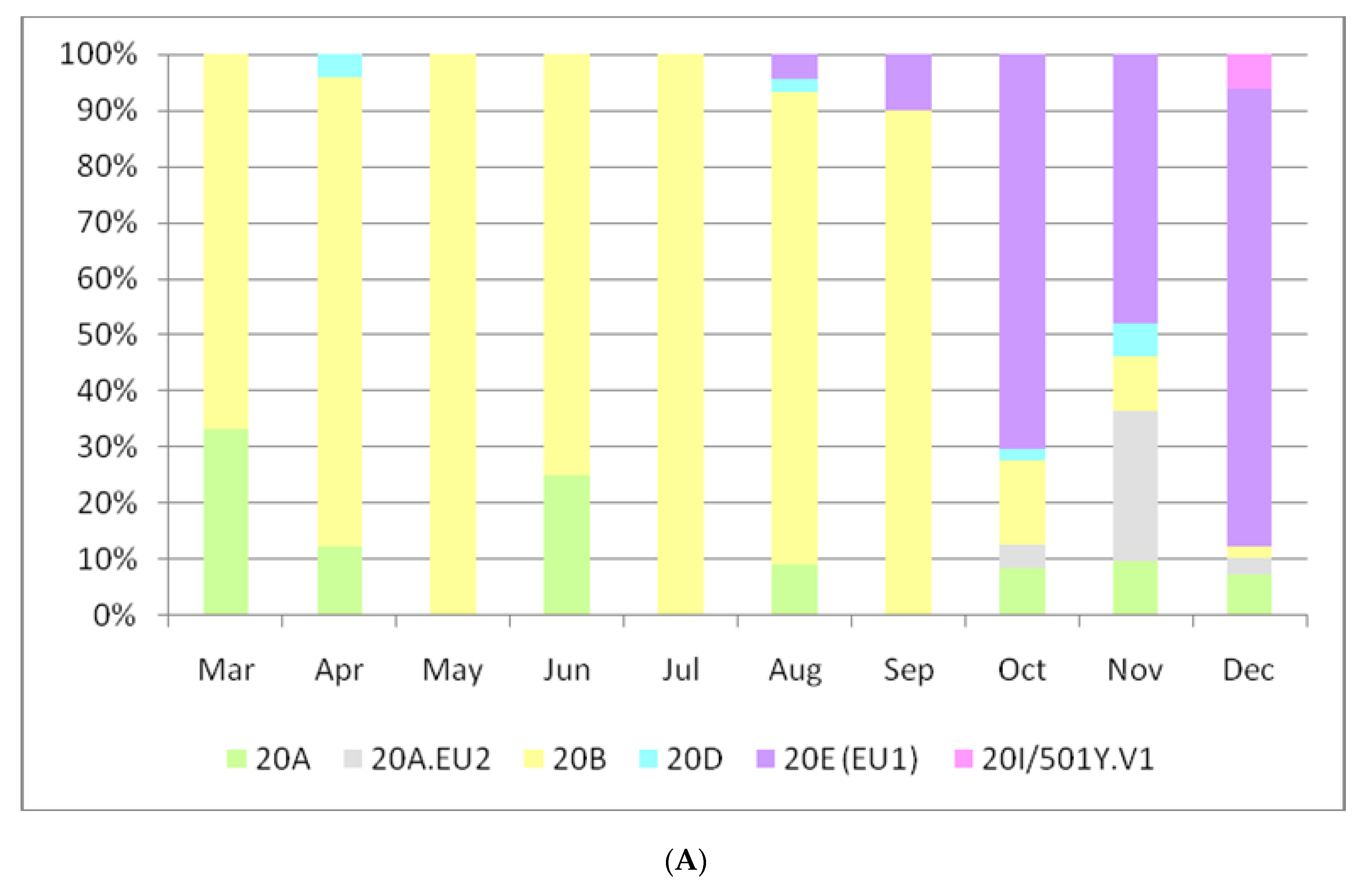
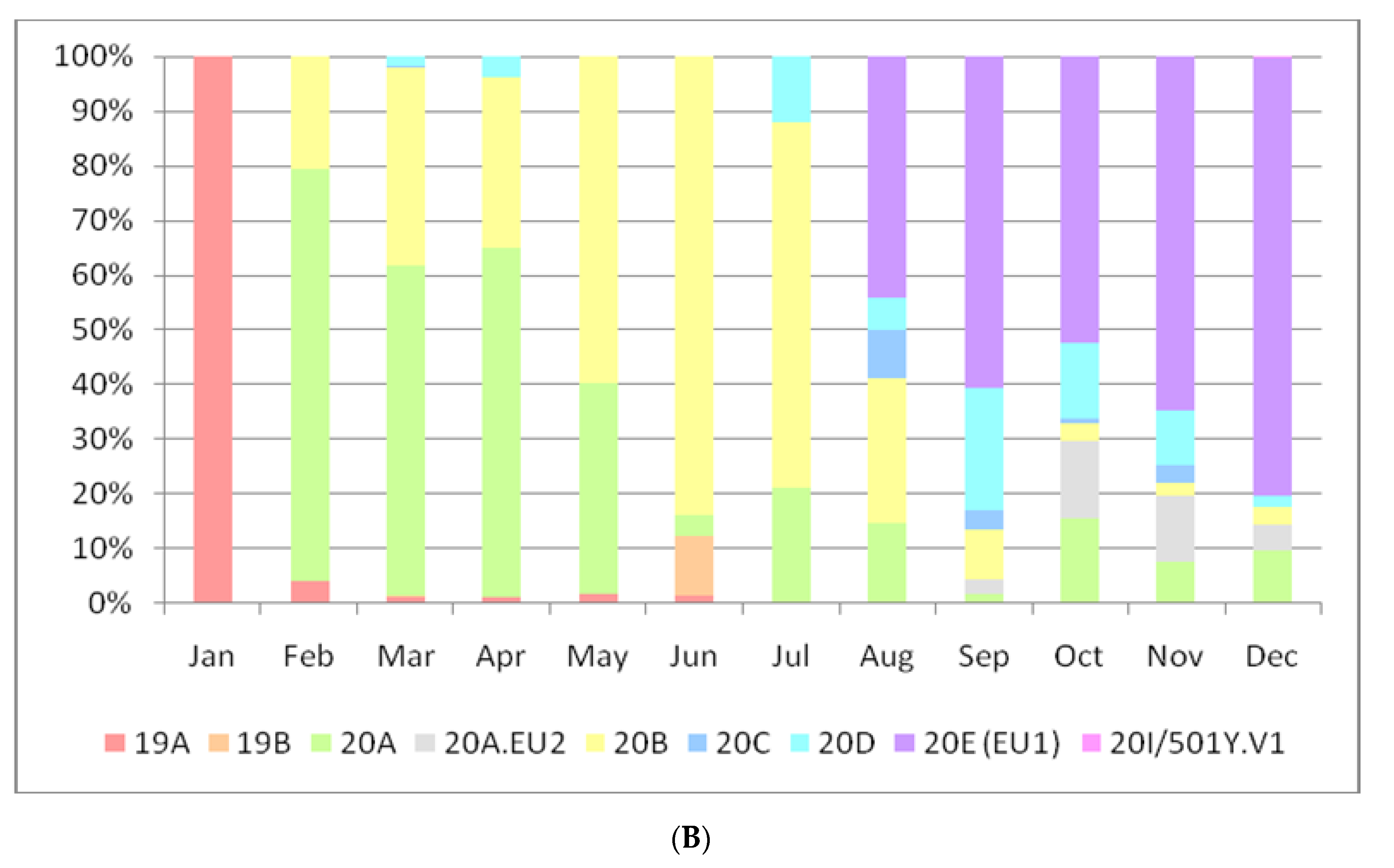
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus Investigating and Research Team. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 14 February 2021).
- Giovanetti, M.; Angeletti, S.; Benvenuto, D.; Ciccozzi, M. A Doubt of Multiple Introduction of SARS-CoV-2 in Italy: A Preliminary Overview. J. Med. Virol. 2020, 92, 1634–1636. [Google Scholar] [CrossRef]
- Stefanelli, P.; Faggioni, G.; Lo Presti, A.; Fiore, S.; Marchi, A.; Benedetti, E.; Fabiani, C.; Anselmo, A.; Ciammaruconi, A.; Fortunato, A.; et al. Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: Additional clues on multiple introductions and further circulation in Europe. Euro. Surveill. 2020, 25. [Google Scholar] [CrossRef] [Green Version]
- Capobianchi, M.R.; Rueca, M.; Messina, F.; Giombini, E.; Carletti, F.; Colavita, F.; Castilletti, C.; Lalle, E.; Bordi, L.; Vairo, F.; et al. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin. Microbiol. Infect. 2020, 26, 954–956. [Google Scholar] [CrossRef]
- Alteri, C.; Cento, V.; Piralla, A.; Costabile, V.; Tallarita, M.; Colagrossi, L.; Renica, S.; Giardina, F.; Novazzi, F.; Gaiarsa, S.; et al. Genomic Epidemiology of SARS-CoV-2 Reveals Multiple Lineages and Early Spread of SARS-CoV-2 Infections in Lombardy, Italy. medRxiv 2020. [Google Scholar] [CrossRef]
- Micheli, V.; Rimoldi, S.; Francesca, R.; Comandatore, F.; Mancon, A.; Gigantiello, A.; Brilli, M.; Mileto, D.; Pagani, C.; Lombardi, A.; et al. Geographic Reconstruction of the SARS-CoV-2 Outbreak in Lombardy (Italy) during the Early Phase. J. Med. Virol. 2020, 93, 1752–1757. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Map. Available online: https://coronavirus.jhu.edu/map.html (accessed on 14 February 2021).
- GISAID-Clade and Lineage Nomenclature Aids in Genomic Epidemiology of Active hCoV-19 Viruses. Available online: https://www.gisaid.org/references/statements-clarifications/clade-and-lineage-nomenclature-aids-in-genomic-epidemiology-of-active-hcov-19-viruses/ (accessed on 14 February 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Hadfield, J.; Neher, R.A.; Bedford, T. Year-letter Genetic Clade Naming for SARS-CoV-2 on Nextstrain.org. Available online: https://nextstrain.org//blog/2020-06-02-SARSCoV2-clade-naming (accessed on 14 February 2021).
- Updated Nextstain SARS-CoV-2 Clade Naming Strategy-SARS-CoV-2 Coronavirus/nCoV-2019 Genomic Epidemiology. Available online: https://virological.org/t/updated-nextstain-sars-cov-2-clade-naming-strategy/581 (accessed on 15 February 2021).
- Risk Assessment: Risk Related to Spread of New SARS-CoV-2 Variants of Concern in the EU/EEA. Available online: https://www.ecdc.europa.eu/en/publications-data/covid-19-risk-assessment-spread-new-sars-cov-2-variants-eueea (accessed on 14 February 2021).
- artic-network/artic-ncov2019. Available online: https://github.com/artic-network/artic-ncov2019 (accessed on 14 February 2021).
- Bianco, A.; Capozzi, L.; Monno, M.R.; Del Sambro, L.; Manzulli, V.; Pesole, G.; Loconsole, D.; Parisi, A. Characterization of Bacillus Cereus Group Isolates from Human Bacteremia by Whole-Genome Sequencing. Front. Microbiol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-Generation Sequencing Alignment Data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef] [PubMed]
- Argimón, S.; Abudahab, K.; Goater, R.J.E.; Fedosejev, A.; Bhai, J.; Glasner, C.; Feil, E.J.; Holden, M.T.G.; Yeats, C.A.; Grundmann, H.; et al. Microreact: Visualizing and Sharing Data for Genomic Epidemiology and Phylogeography. Microb. Genom. 2016, 2, e000093. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v4: Recent Updates and New Developments. Nucleic. Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Khafaie, M.A.; Rahim, F. Cross-Country Comparison of Case Fatality Rates of COVID-19/SARS-COV-2. Osong Public Health Res. Perspect. 2020, 11, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Lu, X.; Chen, Q.; Xu, K.; Chen, Y.; Cheng, L.; Liu, F.; Wu, Z.; Wu, H.; Jin, C.; et al. Patient-Derived Mutations Impact Pathogenicity of SARS-CoV-2. medRxiv 2020. [Google Scholar] [CrossRef]
- Kifer, D.; Bugada, D.; Villar-Garcia, J.; Gudelj, I.; Menni, C.; Sudre, C.; Vučković, F.; Ugrina, I.; Lorini, L.F.; Posso, M.; et al. Effects of Environmental Factors on Severity and Mortality of COVID-19. Front. Med. (Lausanne) 2021, 7, 607786. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, G.; Grassi, G.; Borghi, C.; Ferri, C.; Salvetti, M.; Volpe, M.; SARS-RAS Investigators. Age and Multimorbidity Predict Death Among COVID-19 Patients: Results of the SARS-RAS Study of the Italian Society of Hypertension. Hypertension 2020, 76, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Comas, I.; Candelas, F.G.; Consortium, S.-S.; Stadler, T.; Neher, R.A. Emergence and Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020. medRxiv 2020. [Google Scholar] [CrossRef]
- Guthrie, J.L.; Teatero, S.; Zittermann, S.; Chen, Y.; Sullivan, A.; Rilkoff, H.; Joshi, E.; Sivaraman, K.; de Borja, R.; Sundaravadanam, Y.; et al. Detection of the Novel SARS-CoV-2 European Lineage, B.1.177 in Ontario, Canada. medRxiv 2020. [Google Scholar] [CrossRef]
- Respiratory Viruses SENTINELLE Newsletter. Available online: https://lns.lu/wp-content/uploads/2020/11/weekly_report_main_sem45.html (accessed on 14 February 2021).
- NERVTAG Meeting on SARS-CoV-2 Variant under Investigation VUI-202012/01; New and Emerging Respiratory Virus Threats Advisory Group: London, UK, 2020.
- WHO|SARS-CoV-2 Variant–United Kingdom of Great Britain and Northern Ireland. Available online: http://www.who.int/csr/don/21-december-2020-sars-cov2-variant-united-kingdom/en/ (accessed on 14 February 2021).
- Lenoci, G.; Galante, D.; Ceci, E.; Manzulli, V.; Moramarco, A.M.; Chiaromonte, A.; Labarile, G.; Lattarulo, S.; Resta, A.; Pace, L.; et al. Sars-CoV-2 Isolation from a 10-Day-Old Newborn in Italy: A Case Report. IDCases 2020, 22, e00960. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Lei, Z.; Hai, Z.; Xiao, S.; Rui, J.; Yang, H.; Jing, X.; Wang, H.; Xie, Z.; Luo, P.; et al. Transmission of SARS-CoV-2 in Public Transportation Vehicles: A Case Study in Hunan Province, China. Open Forum. Infect. Dis. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, C.; Dong, H.; Wang, Z.; Martinez, L.; Sun, Z.; Handel, A.; Chen, Z.; Chen, E.; Ebell, M.H.; et al. Community Outbreak Investigation of SARS-CoV-2 Transmission Among Bus Riders in Eastern China. JAMA Intern. Med. 2020, 180, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Comandatore, F.; Chiodi, A.; Gabrieli, P.; Biffignandi, G.B.; Perini, M.; Ricagno, S.; Mascolo, E.; Petazzoni, G.; Ramazzotti, M.; Rimoldi, S.G.; et al. Insurgence and Worldwide Diffusion of Genomic Variants in SARS-CoV-2 Genomes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chinazzi, M.; Davis, J.T.; Ajelli, M.; Gioannini, C.; Litvinova, M.; Merler, S.; Pastore, Y.; Piontti, A.; Mu, K.; Rossi, L.; et al. The Effect of Travel Restrictions on the Spread of the 2019 Novel Coronavirus (COVID-19) Outbreak. Science 2020, 368, 395–400. [Google Scholar] [CrossRef] [Green Version]
- Wells, C.R.; Sah, P.; Moghadas, S.M.; Pandey, A.; Shoukat, A.; Wang, Y.; Wang, Z.; Meyers, L.A.; Singer, B.H.; Galvani, A.P. Impact of International Travel and Border Control Measures on the Global Spread of the Novel 2019 Coronavirus Outbreak. Proc. Natl. Acad. Sci. USA 2020, 117, 7504–7509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Hu, Y.; Chen, L.; Yau, T.; Tong, Y.; Hu, J.; Cai, J.; Chan, K.-H.; Dou, Y.; Deng, J.; et al. Mining of Epitopes on Spike Protein of SARS-CoV-2 from COVID-19 Patients. Cell Res. 2020, 30, 702–704. [Google Scholar] [CrossRef] [PubMed]
- COG-UK. Report on SARS-CoV-2 Spike mutations of interest in the UK. Available online: https://www.cogconsortium.uk/wp-content/uploads/2021/01/Report-2_COG-UK_SARS-CoV-2-Mutations.pdf (accessed on 21 January 2021).
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. The Circulating SARS-CoV-2 Spike Variant N439K Maintains Fitness While Evading Antibody-Mediated Immunity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhou, W.; Xu, C.; Wang, P.; Luo, M.; Xu, Z.; Cheng, R.; Jin, X.; Guo, Y.; Xue, G.; Juan, L.; et al. N439K Variant in Spike Protein May Alter the Infection Efficiency and Antigenicity of SARS-CoV-2 Based on Molecular Dynamics Simulation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key Residues of the Receptor Binding Motif in the Spike Protein of SARS-CoV-2 That Interact with ACE2 and Neutralizing Antibodies. Cell. Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, H. The D614G Mutation in the SARS-CoV-2 Spike Protein Reduces S1 Shedding and Increases Infectivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G Variant Exhibits Efficient Replication Ex Vivo and Transmission in Vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Jain, S.; Xiao, X.; Bogdan, P.; Bruck, J. Predicting the Emergence of SARS-CoV-2 Clades. bioRxiv 2020. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capozzi, L.; Bianco, A.; Del Sambro, L.; Simone, D.; Lippolis, A.; Notarnicola, M.; Pesole, G.; Pace, L.; Galante, D.; Parisi, A. Genomic Surveillance of Circulating SARS-CoV-2 in South East Italy: A One-Year Retrospective Genetic Study. Viruses 2021, 13, 731. https://doi.org/10.3390/v13050731
Capozzi L, Bianco A, Del Sambro L, Simone D, Lippolis A, Notarnicola M, Pesole G, Pace L, Galante D, Parisi A. Genomic Surveillance of Circulating SARS-CoV-2 in South East Italy: A One-Year Retrospective Genetic Study. Viruses. 2021; 13(5):731. https://doi.org/10.3390/v13050731
Chicago/Turabian StyleCapozzi, Loredana, Angelica Bianco, Laura Del Sambro, Domenico Simone, Antonio Lippolis, Maria Notarnicola, Graziano Pesole, Lorenzo Pace, Domenico Galante, and Antonio Parisi. 2021. "Genomic Surveillance of Circulating SARS-CoV-2 in South East Italy: A One-Year Retrospective Genetic Study" Viruses 13, no. 5: 731. https://doi.org/10.3390/v13050731
APA StyleCapozzi, L., Bianco, A., Del Sambro, L., Simone, D., Lippolis, A., Notarnicola, M., Pesole, G., Pace, L., Galante, D., & Parisi, A. (2021). Genomic Surveillance of Circulating SARS-CoV-2 in South East Italy: A One-Year Retrospective Genetic Study. Viruses, 13(5), 731. https://doi.org/10.3390/v13050731