
Host Cell Restriction Factors of Bunyaviruses and Viral Countermeasures



Abstract
:1. Introduction
2. Medical Importance of Viruses in the Bunyavirales Order
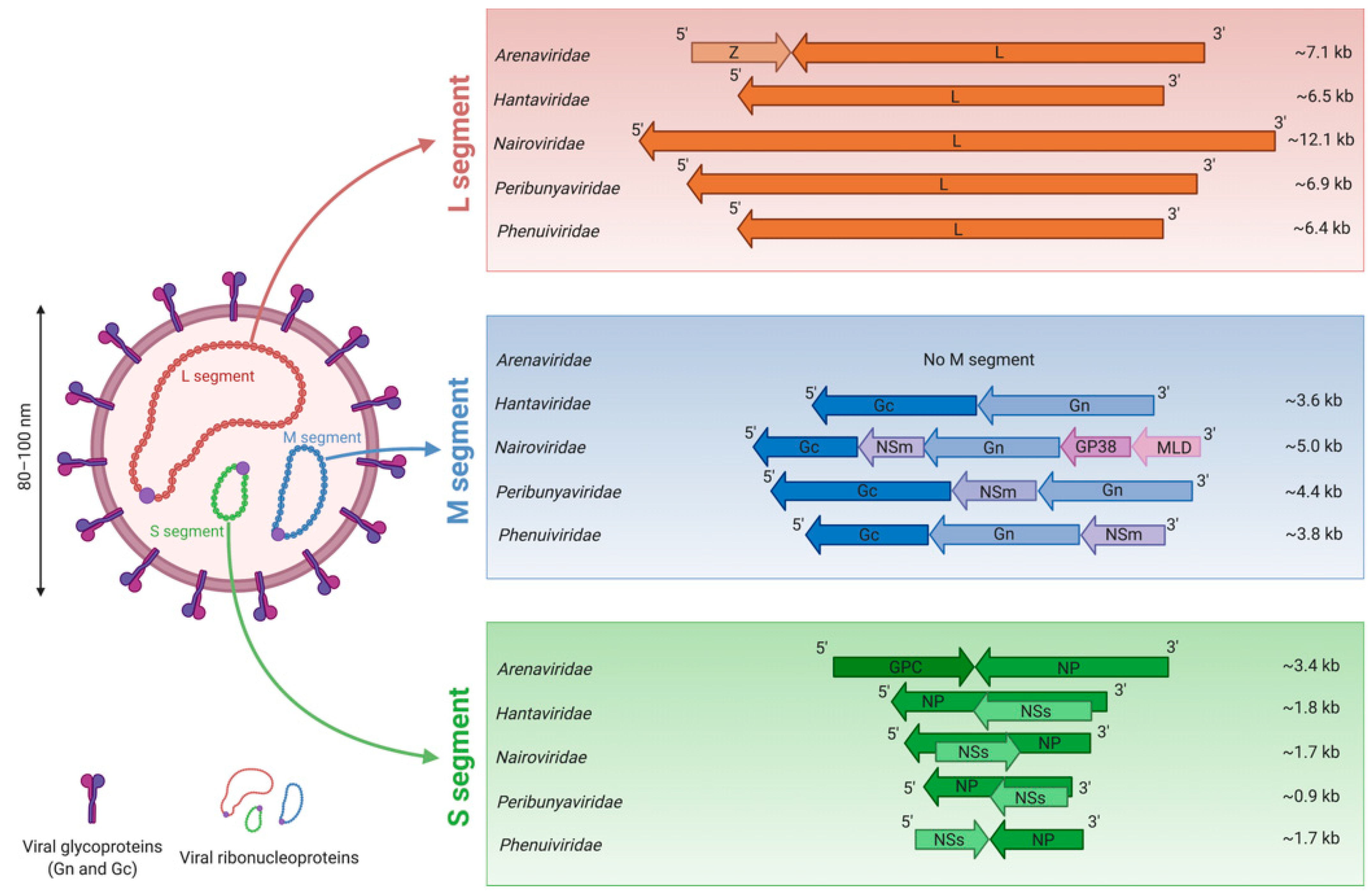
3. Morphology of Bunyavirus Particles and Genome Organization
4. Bunyavirus Cell Cycle
5. Induction of Type I Interferons and Innate Immunity Following Bunyavirus Infection
6. Inhibition of Bunyavirus by IFN-Stimulated Genes and Other Antiviral Factors
6.1. Restriction Factors Targeting Bunyaviruses Entry
6.1.1. IFN-Induced Transmembrane Proteins (IFITMs)
6.1.2. γ-IFN-Inducible Lysosomal Thiol Reductase (GILT)
6.1.3. Cholesterol-25-hydrolase (CH25H)
6.2. Restriction Factors Interfering with Genomic Transcription, Protein Translation and Genomic Replication
6.2.1. Myxovirus Resistance Protein A (MxA)
6.2.2. Protein Kinase R (PKR)
6.2.3. IFN-Induced Protein with Tetratricopeptide Repeats 1 (IFIT1)
6.2.4. Long Isoform of Mono-ADP-ribosyltransferase 12 (PARP12L)
6.2.5. Decapping Protein 2 (Dcp2)
6.2.6. DDX17
6.2.7. IFN-Induced Protein 44 (IFI44)
6.2.8. Promyelocytic Leukemia Protein (PML)
6.2.9. Moloney Leukemia Virus 10 Protein (MOV10)
6.2.10. IFN-Stimulated Gene 20 (ISG20)
6.2.11. 2′-5′-Oligoadenylate Synthetase (2′-5′-OAS)/RNase L
6.3. Restrictions Factors Blocking Viral Assembly and Egress
6.3.1. Viperin
6.3.2. Tetherin
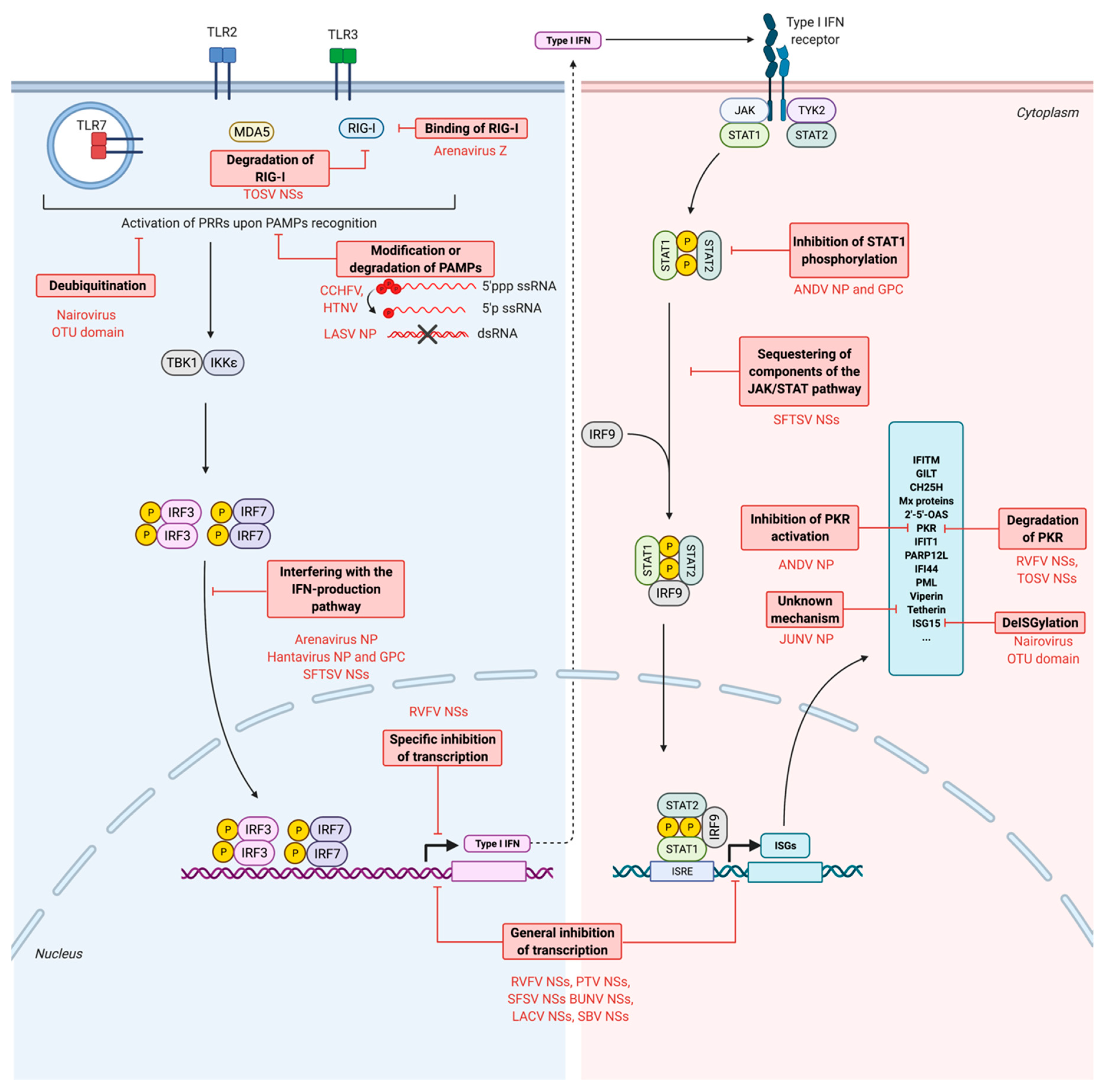
7. Escape of Antiviral Responses by Bunyaviruses
7.1. Escape from the PRRs Recognition
7.1.1. Modification of PAMPs: Processing of the Genome
7.1.2. Degradation of PAMPs
7.2. Inhibition of the Production of IFNs
7.2.1. Global Cellular Transcriptional and Translational Shutoff
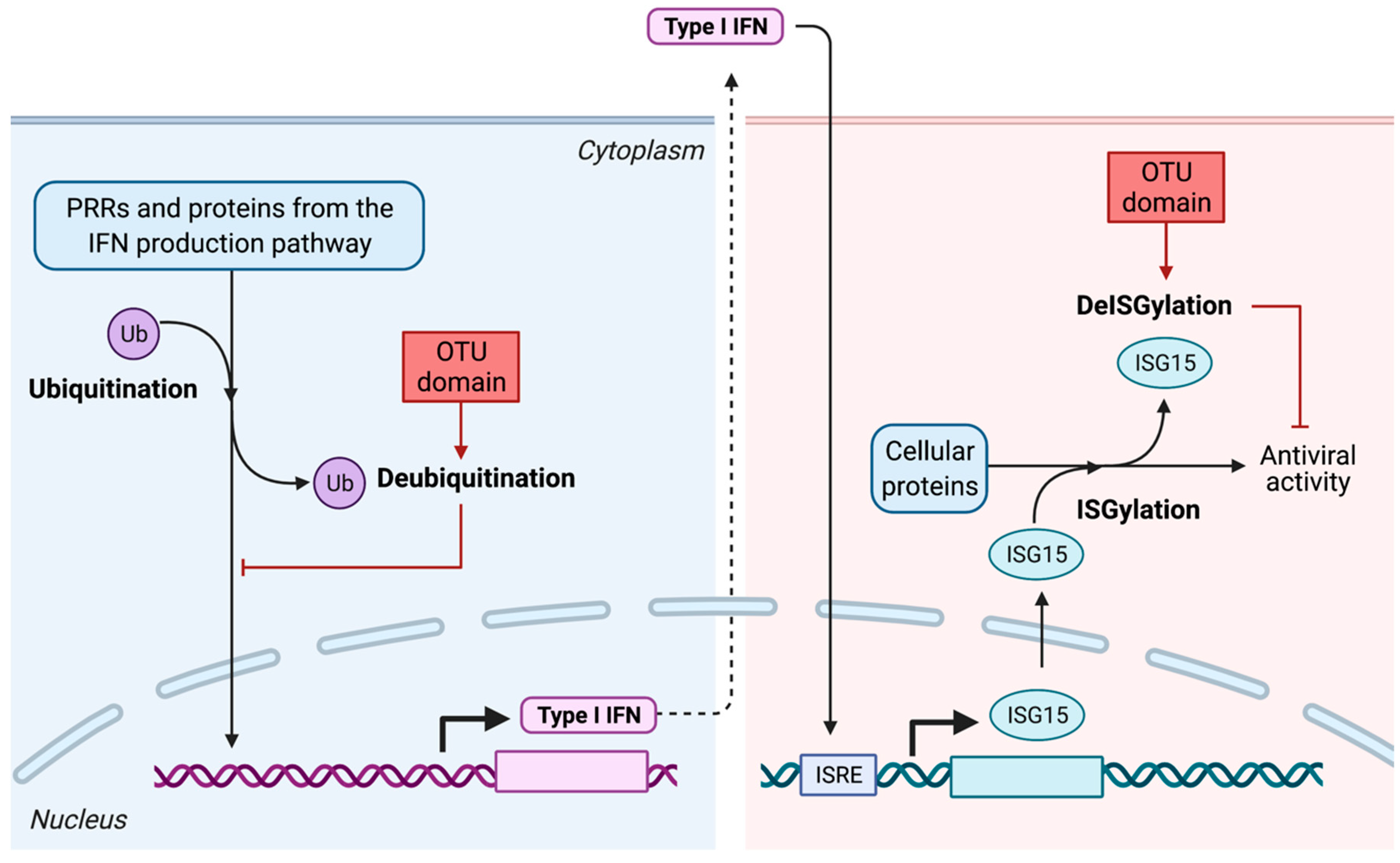
7.2.2. Interference with the IFN Production Pathway
- Alteration of post-translational modifications of key factors from the IFN production pathway: deubiquitination and deISGylation
- Inhibition of the activation of RIG-I
- Interference with the IFN production pathway
7.3. Inhibition of the Production of ISGs
7.4. Inhibition of the Activity of Restriction Factors
7.4.1. Inhibition of PKR
7.4.2. Inhibition of Tetherin
8. Concluding Remarks
Dedication
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; et al. Taxonomy of the Order Bunyavirales: Update 2019. Arch. Virol. 2019, 164, 1949–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linero, F.N.; Sepúlveda, C.S.; Giovannoni, F.; Castilla, V.; García, C.C.; Scolaro, L.A.; Damonte, E.B. Host Cell Factors as Antiviral Targets in Arenavirus Infection. Viruses 2012, 4, 1569–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, R.M.; Weber, F. Bunyaviruses and the Type I Interferon System. Viruses 2009, 1, 1003–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasecka, L.; Baron, M.D. The Molecular Biology of Nairoviruses, an Emerging Group of Tick-borne Arboviruses. Arch. Virol. 2014, 159, 1249–1265. [Google Scholar] [CrossRef] [PubMed]
- Eifan, S.; Schnettler, E.; Dietrich, I.; Kohl, A.; Blomström, A.-L. Non-Structural Proteins of Arthropod-Borne Bunyaviruses: Roles and Functions. Viruses 2013, 5, 2447–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuerth, J.D.; Weber, F. Phleboviruses and the Type I Interferon Response. Viruses 2016, 8, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prioritizing Diseases for Research and Development in Emergency Contexts. Available online: https://www.who.int/activities/prioritizing-diseases-for-research-and-development-in-emergency-contexts (accessed on 16 March 2021).
- Hughes, H.R.; Adkins, S.; Alkhovskiy, S.; Beer, M.; Blair, C.; Calisher, C.H.; Drebot, M.; Lambert, A.J.; De Souza, W.M.; Marklewitz, M.; et al. ICTV Virus Taxonomy Profile: Peribunyaviridae. J. Gen. Virol. 2020, 101, 1–2. [Google Scholar] [CrossRef]
- Garrison, A.R.; Alkhovsky, С.; Avšič-Županc, T.; Bente, D.A.; Bergeron, É.; Burt, F.; Di Paola, N.; Ergünay, K.; Hewson, R.; Kuhn, J.H.; et al. ICTV Virus Taxonomy Profile: Nairoviridae. J. Gen. Virol. 2020, 101, 798–799. [Google Scholar] [CrossRef]
- Walter, C.T.; Barr, J.N. Recent Advances in the Molecular and Cellular Biology of Bunyaviruses. J. Gen. Virol. 2011, 92, 2467–2484. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Buchmeier, M.J.; Charrel, R.N.; Clegg, J.C.S.; Gonzalez, J.-P.J.; Günther, S.; Hepojoki, J.; Kuhn, J.H.; Lukashevich, I.S.; Romanowski, V.; et al. ICTV Virus Taxonomy Profile: Arenaviridae. J. Gen. Virol. 2019, 100, 1200–1201. [Google Scholar] [CrossRef]
- Schreur, P.J.W.; Kormelink, R.; Kortekaas, J. Genome Packaging of the Bunyavirales. Curr. Opin. Virol. 2018, 33, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Muyangwa, M.; Martynova, E.V.; Khaiboullina, S.F.; Morzunov, S.P.; Rizvanov, A.A. Hantaviral Proteins: Structure, Functions, and Role in Hantavirus Infection. Front. Microbiol. 2015, 6, 1326. [Google Scholar] [CrossRef] [PubMed]
- Albariño, C.G.; Bird, B.H.; Nichol, S.T. A Shared Transcription Termination Signal on Negative and Ambisense RNA Genome Segments of Rift Valley Fever, Sandfly Fever Sicilian, and Toscana Viruses. J. Virol. 2007, 81, 5246–5256. [Google Scholar] [CrossRef] [Green Version]
- Barnwal, B.; Karlberg, H.; Mirazimi, A.; Tan, Y.-J. The Non-structural Protein of Crimean-Congo Hemorrhagic Fever Virus Disrupts the Mitochondrial Membrane Potential and Induces Apoptosis. J. Biol. Chem. 2016, 291, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zivcec, M.; Scholte, F.E.M.; Spiropoulou, C.F.; Spengler, J.R.; Bergeron, É. Molecular Insights into Crimean-Congo Hemorrhagic Fever Virus. Viruses 2016, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinschewer, D.D.; Pérez, M.; De La Torre, J.C. Dual Role of the Lymphocytic Choriomeningitis Virus Intergenic Region in Transcription Termination and Virus Propagation. J. Virol. 2005, 79, 4519–4526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olschewski, S.; Cusack, S.; Rosenthal, M. The Cap-Snatching Mechanism of Bunyaviruses. Trends Microbiol. 2020, 28, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat. Rev. Immunol. 2020, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Gale, M. Differential Recognition of Double-stranded RNA by RIG-I–like Receptors in Antiviral Immunity. J. Exp. Med. 2008, 205, 1523–1527. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Kolokoltsova, O.A.; Yun, N.E.; Seregin, A.V.; Poussard, A.L.; Walker, A.G.; Brasier, A.R.; Zhao, Y.; Tian, B.; De La Torre, J.C.; et al. Junín Virus Infection Activates the Type I Interferon Pathway in a RIG-I-Dependent Manner. PLoS Negl. Trop. Dis. 2012, 6, e1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, Y.-Q.; Ning, Y.-J.; Wang, H.; Deng, F. A RIG-I–like Receptor Directs Antiviral Responses to a Bunyavirus and is Antagonized by Virus-induced Blockade of TRIM25-mediated Ubiquitination. J. Biol. Chem. 2020, 295, 9691–9711. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Andersson, I.; Klingström, J.; Schümann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Mühlberger, E.; et al. Processing of Genome 5′ Termini as a Strategy of Negative-Strand RNA Viruses to Avoid RIG-I-Dependent Interferon Induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Gawanbacht, A.; Habjan, M.; Rang, A.; Borner, C.; Schmidt, A.M.; Veitinger, S.; Jacob, R.; Devignot, S.; Kochs, G.; et al. Incoming RNA Virus Nucleocapsids Containing a 5′-Triphosphorylated Genome Activate RIG-I and Antiviral Signaling. Cell Host Microbe 2013, 13, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-H.; Lalwani, P.; Raftery, M.J.; Matthaei, M.; Lütteke, N.; Kirsanovs, S.; Binder, M.; Ulrich, R.G.; Giese, T.; Wolff, T.; et al. RNA Helicase Retinoic Acid-inducible Gene I as a Sensor of Hantaan Virus Replication. J. Gen. Virol. 2011, 92, 2191–2200. [Google Scholar] [CrossRef] [PubMed]
- Spengler, J.R.; Patel, J.R.; Chakrabarti, A.K.; Zivcec, M.; García-Sastre, A.; Spiropoulou, C.F.; Bergeron, É. RIG-I Mediates an Antiviral Response to Crimean-Congo Hemorrhagic Fever Virus. J. Virol. 2015, 89, 10219–10229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and Inhibition of Type I Interferon Responses by Distinct Components of Lymphocytic Choriomeningitis Virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handke, W.; Oelschlegel, R.; Franke, R.; Krüger, D.H.; Rang, A. Hantaan Virus Triggers TLR3-Dependent Innate Immune Responses. J. Immunol. 2009, 182, 2849–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macal, M.; Lewis, G.M.; Kunz, S.; Flavell, R.; Harker, J.A.; Zúñiga, E.I. Plasmacytoid Dendritic Cells Are Productively Infected and Activated through TLR-7 Early after Arenavirus Infection. Cell Host Microbe 2012, 11, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, C.D.; Lavanya, M.; Wang, E.; Ross, S.R. Junin Virus Infects Mouse Cells and Induces Innate Immune Responses. J. Virol. 2011, 85, 11058–11068. [Google Scholar] [CrossRef] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Garrison, A.R.; Smith, D.R.; Golden, J.W. Animal Models for Crimean-Congo Hemorrhagic Fever Human Disease. Viruses 2019, 11, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouloy, M.; Janzen, C.; Vialat, P.; Khun, H.; Pavlovic, J.; Huerre, M.; Haller, O. Genetic Evidence for an Interferon-Antagonistic Function of Rift Valley Fever Virus Nonstructural Protein NSs. J. Virol. 2001, 75, 1371–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendenhall, M.; Wong, M.-H.; Skirpstunas, R.; Morrey, J.D.; Gowen, B.B. Punta Toro Virus (Bunyaviridae, Phlebovirus) Infection in Mice: Strain Differences in Pathogenesis and Host Interferon Response. Virology 2009, 395, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakqori, G.; Delhaye, S.; Habjan, M.; Blair, C.D.; Sánchez-Vargas, I.; Olson, K.E.; Attarzadeh-Yazdi, G.; Fragkoudis, R.; Kohl, A.; Kalinke, U.; et al. La Crosse Bunyavirus Nonstructural Protein NSs Serves to Suppress the Type I Interferon System of Mammalian Hosts. J. Virol. 2007, 81, 4991–4999. [Google Scholar] [CrossRef] [Green Version]
- Boyd, A.; Fazakerley, J.K.; Bridgen, A. Pathogenesis of Dugbe Virus Infection in Wild-type and Interferon-deficient Mice. J. Gen. Virol. 2006, 87, 2005–2009. [Google Scholar] [CrossRef]
- Hefti, H.P.; Frese, M.; Landis, H.; Di Paolo, C.; Aguzzi, A.; Haller, O.; Pavlovic, J. Human MxA Protein Protects Mice Lacking a Functional Alpha/Beta Interferon System against La Crosse Virus and Other Lethal Viral Infections. J. Virol. 1999, 73, 6984–6991. [Google Scholar] [CrossRef] [Green Version]
- Wichmann, D.; Grone, H.-J.; Frese, M.; Pavlovic, J.; Anheier, B.; Haller, O.; Klenk, H.-D.; Feldmann, H. Hantaan Virus Infection Causes an Acute Neurological Disease That Is Fatal in Adult Laboratory Mice. J. Virol. 2002, 76, 8890–8899. [Google Scholar] [CrossRef] [Green Version]
- Kell, A.M.; Hemann, E.A.; Turnbull, J.B.; Gale, M. RIG-I-like Receptor Activation Drives Type I IFN and Antiviral Signaling to Limit Hantaan Orthohantavirus Replication. PLoS Pathog. 2020, 16, e1008483. [Google Scholar] [CrossRef] [Green Version]
- Hawman, D.W.; Meade-White, K.; Leventhal, S.; Feldmann, F.; Okumura, A.; Smith, B.; Scott, D.; Feldmann, H. Immunocompetent Mouse Model for Crimean-Congo Hemorrhagic Fever Virus. eLife 2021, 10. [Google Scholar] [CrossRef]
- Pinkham, C.; Dahal, B.; De La Fuente, C.L.; Bracci, N.; Beitzel, B.; Lindquist, M.; Garrison, A.; Schmaljohn, C.; Palacios, G.; Narayanan, A.; et al. Alterations in the Host Transcriptome in vitro Following Rift Valley Fever Virus Infection. Sci. Rep. 2017, 7, 14385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havranek, K.E.; White, L.A.; Lanchy, J.-M.; Lodmell, J.S. Transcriptome Profiling in Rift Valley Fever Virus Infected Cells Reveals Modified Transcriptional and Alternative Splicing Programs. PLoS ONE 2019, 14, e0217497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomström, A.-L.; Gu, Q.; Barry, G.; Wilkie, G.; Skelton, J.K.; Baird, M.; McFarlane, M.; Schnettler, E.; Elliott, R.M.; Palmarini, M.; et al. Transcriptome Analysis Reveals the Host Response to Schmallenberg Virus in Bovine Cells and Antagonistic Effects of the NSs Protein. BMC Genom. 2015, 16, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, S.; Yen, J.Y.; Honko, A.N.; Garamszegi, S.; Caballero, I.S.; Johnson, J.C.; Mucker, E.M.; Trefry, J.C.; Hensley, L.E.; Connor, J.H. Transcriptional Profiling of the Circulating Immune Response to Lassa Virus in an Aerosol Model of Exposure. PLoS Negl. Trop. Dis. 2013, 7, e2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapata, J.C.; Carrion, R.; Patterson, J.L.; Crasta, O.; Zhang, Y.; Mani, S.; Jett, M.; Poonia, B.; Djavani, M.; White, D.M.; et al. Transcriptome Analysis of Human Peripheral Blood Mononuclear Cells Exposed to Lassa Virus and to the Attenuated Mopeia/Lassa Reassortant 29 (ML29), a Vaccine Candidate. PLoS Negl. Trop. Dis. 2013, 7, e2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, M.S.; Farzan, M. The Broad-spectrum Antiviral Functions of IFIT and IFITM Proteins. Nat. Rev. Immunol. 2012, 13, 46–57. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Winkler, C.A.; An, P.; Guo, J.-T. IFITM Genes, Variants, and Their Roles in the Control and Pathogenesis of Viral Infections. Front. Microbiol. 2019, 9, 3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Radoshitzky, S.R.; Kota, K.P.; Altamura, L.A.; Smith, J.M.; Packard, B.Z.; Kuhn, J.H.; Costantino, J.; et al. IFITM-2 and IFITM-3 but Not IFITM-1 Restrict Rift Valley Fever Virus. J. Virol. 2013, 87, 8451–8464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu-Yang, Z.; Pei-Yu, B.; Chuan-Tao, Y.; Wei, Y.; Hong-Wei, M.; Kang, T.; Chun-Mei, Z.; Ying-Feng, L.; Xin, W.; Ping-Zhong, W.; et al. Interferon-Induced Transmembrane Protein 3 Inhibits Hantaan Virus Infection, and Its Single Nucleotide Polymorphism rs12252 Influences the Severity of Hemorrhagic Fever with Renal Syndrome. Front. Immunol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jegado, B.; Journo, C.; Mahieux, R. Un Double Effet Antiviral des IFITM sur les Virus Enveloppés. Médecine Sci. 2018, 34, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wickenhagen, A.; Turnbull, M.L.; Rezelj, V.V.; Kreher, F.; Tilston-Lunel, N.L.; Slack, G.S.; Brennan, B.; Koudriakova, E.; Shaw, A.E.; et al. Interferon-Stimulated Gene (ISG)-Expression Screening Reveals the Specific Antibunyaviral Activity of ISG20. J. Virol. 2018, 92, e02140–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; Van Der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Chi, X.; Wei, H.; Chen, Y.; Chen, Z.; Huang, S.; Chen, J.-L. Influenza A Virus-Induced Degradation of Eukaryotic Translation Initiation Factor 4B Contributes to Viral Replication by Suppressing IFITM3 Protein Expression. J. Virol. 2014, 88, 8375–8385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Lam, T.H.; Soh, M.K.; Ye, Z.; Chen, J.; Ren, E.C. Influenza A Virus Facilitates Its Infectivity by Activating p53 to Inhibit the Expression of Interferon-Induced Transmembrane Proteins. Front. Immunol. 2018, 9, 1193. [Google Scholar] [CrossRef]
- Chen, D.; Hou, Z.; Jiang, D.; Zheng, M.; Li, G.; Zhang, Y.; Li, R.; Lin, H.; Chang, J.; Zeng, H.; et al. GILT Restricts the Cellular Entry Mediated by the Envelope Glycoproteins of SARS-CoV, Ebola Virus and Lassa Fever Virus. Emerg. Microbes Infect. 2019, 8, 1511–1523. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Chen, J.; Li, M.; Chen, M.; Sun, C. Multifaceted Functions of CH25H and 25HC to Modulate the Lipid Metabolism, Immune Responses, and Broadly Antiviral Activities. Viruses 2020, 12, 727. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-Inducible Cholesterol-25-Hydroxylase Broadly Inhibits Viral Entry by Production of 25-Hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava-Ranjan, P.; Bergeron, E.; Chakrabarti, A.K.; Albariño, C.G.; Flint, M.; Nichol, S.T.; Spiropoulou, C.F. 25-Hydroxycholesterol Inhibition of Lassa Virus Infection through Aberrant GP1 Glycosylation. MBio 2016, 7, e01808-16. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.; Johansson, C.; Mirazimi, A. Crimean-Congo Hemorrhagic Fever Virus Entry and Replication is Clathrin-, pH- and Cholesterol-dependent. J. Gen. Virol. 2009, 90, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R.G. Interferon-inducible Antiviral Effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Frese, M.; Kochs, G.; Feldmann, H.; Hertkorn, C.; Haller, O. Inhibition of Bunyaviruses, Phleboviruses, and Hantaviruses by Human MxA Protein. J. Virol. 1996, 70, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichelt, M.; Stertz, S.; Krijnse-Locker, J.; Haller, O.; Kochs, G. Missorting of LaCrosse Virus Nucleocapsid Protein by the Interferon-induced MxA GTPase Involves Smooth ER Membranes. Traffic 2004, 5, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Janzen, C.; Hohenberg, H.; Haller, O. Antivirally Active MxA Protein Sequesters La Crosse Virus Nucleocapsid Protein into Perinuclear Complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 3153–3158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, T.A.; Carlson, J.O.; Beaty, B.J.; Bowen, R.A.; Olson, K.E. Expression of Human MxA Protein in Mosquito Cells Interferes with LaCrosse Virus Replication. J. Virol. 2001, 75, 3001–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandrock, M.; Frese, M.; Haller, O.; Kochs, G. Interferon-Induced Rat Mx Proteins Confer Resistance to Rift Valley Fever Virus and Other Arthropod-Borne Viruses. J. Interf. Cytokine Res. 2001, 21, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Andersson, I.; Bladh, L.; Mousavi-Jazi, M.; Magnusson, K.-E.; Lundkvist, A.; Haller, O.; Mirazimi, A. Human MxA Protein Inhibits the Replication of Crimean-Congo Hemorrhagic Fever Virus. J. Virol. 2004, 78, 4323–4329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgen, A.; Dalrymple, D.A.; Weber, F.; Elliott, R.M. Inhibition of Dugbe Nairovirus Replication by Human MxA Protein. Virus Res. 2004, 99, 47–50. [Google Scholar] [CrossRef]
- Nam, J.-H.; Hwang, K.-A.; Yu, C.-H.; Kang, T.-H.; Shin, J.-Y.; Choi, W.-Y.; Kim, I.-B.; Joo, Y.-R.; Cho, H.-W.; Park, K.-Y. Expression of Interferon Inducible Genes Following Hantaan Virus Infection as a Mechanism of Resistance in A549 Cells. Virus Genes 2003, 26, 31–38. [Google Scholar] [CrossRef]
- Khaiboullina, S.F.; Rizvanov, A.A.; Otteson, E.; Miyazato, A.; Maciejewski, J.; Jeor, S.S. Regulation of Cellular Gene Expression in Endothelial Cells by Sin Nombre and Prospect Hill Viruses. Viral Immunol. 2004, 17, 234–251. [Google Scholar] [CrossRef]
- Khaiboullina, S.F.; Rizvanov, A.A.; Deyde, V.M.; Jeor, S.C.S. Andes Virus Stimulates Interferon-inducible MxA Protein Expression in Endothelial Cells. J. Med. Virol. 2004, 75, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, M.; Melén, K.; Vaheri, A.; Julkunen, I. Inhibition of Puumala and Tula Hantaviruses in Vero Cells by MxA Protein. Virology 1996, 224, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oelschlegel, R.; Krüger, D.H.; Rang, A. MxA-independent Inhibition of Hantaan Virus Replication Induced by Type I and Type II Interferon in vitro. Virus Res. 2007, 127, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Graf, L.; Dick, A.; Sendker, F.; Barth, E.; Marz, M.; Daumke, O.; Kochs, G. Effects of Allelic Variations in the Human Myxovirus Resistance Protein A on Its Antiviral Activity. J. Biol. Chem. 2018, 293, 3056–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habjan, M.; Penski, N.; Wagner, V.; Spiegel, M.; Överby, A.K.; Kochs, G.; Huiskonen, J.T.; Weber, F. Efficient Production of Rift Valley Fever Virus-like Particles: The Antiviral Protein MxA Can Inhibit Primary Transcription of Bunyaviruses. Virology 2009, 385, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Carlton-Smith, C.; Elliott, R.M. Viperin, MTAP44, and Protein Kinase R Contribute to the Interferon-Induced Inhibition of Bunyamwera Orthobunyavirus Replication. J. Virol. 2012, 86, 11548–11557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streitenfeld, H.; Boyd, A.; Fazakerley, J.K.; Bridgen, A.; Elliott, R.M.; Weber, F. Activation of PKR by Bunyamwera Virus Is Independent of the Viral Interferon Antagonist NSs. J. Virol. 2003, 77, 5507–5511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habjan, M.; Pichlmair, A.; Elliott, R.M.; Overby, A.K.; Glatter, T.; Gstaiger, M.; Superti-Furga, G.; Unger, H.; Weber, F. NSs Protein of Rift Valley Fever Virus Induces the Specific Degradation of the Double-Stranded RNA-Dependent Protein Kinase. J. Virol. 2009, 83, 4365–4375. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, T.; Narayanan, K.; Won, S.; Kamitani, W.; Peters, C.J.; Makino, S. Rift Valley Fever Virus NSs Protein Promotes Post-Transcriptional Downregulation of Protein Kinase PKR and Inhibits eIF2α Phosphorylation. PLoS Pathog. 2009, 5, e1000287. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Kolokoltsova, O.A.; Mateer, E.J.; Koma, T.; Paessler, S. Highly Pathogenic New World Arenavirus Infection Activates the Pattern Recognition Receptor Protein Kinase R without Attenuating Virus Replication in Human Cells. J. Virol. 2017, 91, e01090–17. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Lassnig, C.; Eberle, C.-A.; Górna, M.W.; Baumann, C.L.; Burkard, T.R.; Bürckstümmer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an Antiviral Protein That Recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Atasheva, S.; Akhrymuk, M.; Frolova, E.I.; Frolov, I. New PARP Gene with an Anti-Alphavirus Function. J. Virol. 2012, 86, 8147–8160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atasheva, S.; Frolova, E.I.; Frolov, I. Interferon-Stimulated Poly(ADP-Ribose) Polymerases Are Potent Inhibitors of Cellular Translation and Virus Replication. J. Virol. 2014, 88, 2116–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, K.C.; McLane, L.M.; Maqbool, T.; Panda, D.; Gordesky-Gold, B.; Cherry, S. A Genome-wide RNAi Screen Reveals That mRNA Decapping Restricts Bunyaviral Replication by Limiting the Pools of Dcp2-accessible Targets for Cap-snatching. Genes Dev. 2013, 27, 1511–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhou, Y.; Moffett, P. Alterations in Cellular RNA Decapping Dynamics Affect Tomato Spotted Wilt Virus Cap Snatching and Infection in Arabidopsis. New Phytol. 2019, 224, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Elliott, R.M. Non-viral Sequences at the 5’ Ends of Dugbe Nairovirus S mRNAs. J. Gen. Virol. 1993, 74, 2293–2297. [Google Scholar] [CrossRef]
- Decroly, E.; Ferron, F.; Lescar, J.; Canard, B. Conventional and Unconventional Mechanisms for Capping Viral mRNA. Nat. Rev. Genet. 2011, 10, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Moy, R.H.; Cole, B.S.; Yasunaga, A.; Gold, B.; Shankarling, G.; Varble, A.; Molleston, J.M.; Tenoever, B.R.; Lynch, K.W.; Cherry, S. Stem-Loop Recognition by DDX17 Facilitates miRNA Processing and Antiviral Defense. Cell 2014, 158, 764–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.R.; Mrozowich, T.; Park, S.M.; D’Souza, S.; Henrickson, A.; Vigar, J.R.J.; Wieden, H.-J.; Owens, R.J.; Demeler, B.; Patel, T.R. Human DDX17 Unwinds Rift Valley Fever Virus Non-Coding RNAs. Int. J. Mol. Sci. 2020, 22, 54. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nogales, A.; Martinez-Sobrido, L.; Topham, D.J. Interferon-Induced Protein 44 Interacts with Cellular FK506-Binding Protein 5, Negatively Regulates Host Antiviral Responses, and Supports Virus Replication. MBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Djavani, M.; Rodas, J.; Lukashevich, I.S.; Horejsh, D.; Pandolfi, P.P.; Borden, K.L.B.; Salvato, M.S. Role of the Promyelocytic Leukemia Protein PML in the Interferon Sensitivity of Lymphocytic Choriomeningitis Virus. J. Virol. 2001, 75, 6204–6208. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, W.V.; Pinschewer, D.D.; Klenerman, P.; Rousson, V.; Gaboli, M.; Pandolfi, P.P.; Zinkernagel, R.M.; Salvato, M.S.; Hengartner, H. Effects of Promyelocytic Leukemia Protein on Virus-Host Balance. J. Virol. 2002, 76, 3810–3818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borden, K.L.B.; Dwyer, E.J.C.; Salvato, M.S. An Arenavirus RING (Zinc-Binding) Protein Binds the Oncoprotein Promyelocyte Leukemia Protein (PML) and Relocates PML Nuclear Bodies to the Cytoplasm. J. Virol. 1998, 72, 758–766. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Han, Y.; Dang, Y.; Fu, W.; Zhou, T.; Ptak, R.G.; Zheng, Y.-H. Moloney Leukemia Virus 10 (MOV10) Protein Inhibits Retrovirus Replication. J. Biol. Chem. 2010, 285, 14346–14355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Sun, Q.; Liu, Y.; Cen, S.; Zhang, Q. The MOV10 Helicase Restricts Hepatitis B Virus Replication by Inhibiting Viral Reverse Transcription. J. Biol. Chem. 2019, 294, 19804–19813. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.D.; Rice, C.M. A Diverse Range of Gene Products Are Effectors of the Type I Interferon Antiviral Response. Nat. Cell Biol. 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, R.A.; Ghosh, A.; Wallerath, C.; Hornung, V.; Coyne, C.B.; Sarkar, S.N. MOV10 Provides Antiviral Activity against RNA Viruses by Enhancing RIG-I-MAVS-Independent IFN Induction. J. Immunol. 2016, 196, 3877–3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Huang, F.; Tan, L.; Bai, C.; Chen, B.; Liu, J.; Liang, J.; Liu, C.; Zhang, S.; Lu, G.; et al. Host Protein Moloney Leukemia Virus 10 (MOV10) Acts as a Restriction Factor of Influenza A Virus by Inhibiting the Nuclear Import of the Viral Nucleoprotein. J. Virol. 2016, 90, 3966–3980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haussecker, D.; Cao, D.; Huang, Y.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Capped Small RNAs and MOV10 in Human Hepatitis Delta Virus Replication. Nat. Struct. Mol. Biol. 2008, 15, 714–721. [Google Scholar] [CrossRef] [Green Version]
- Mo, Q.; Xu, Z.; Deng, F.; Wang, H.; Ning, Y.-J. Host Restriction of Emerging High-pathogenic Bunyaviruses via MOV10 by Targeting Viral Nucleoprotein and Blocking Ribonucleoprotein Assembly. PLoS Pathog. 2020, 16, e1009129. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, N.; Woodson, S.E.; Dong, Q.; Wang, J.; Liang, Y.; Rijnbrand, R.; Wei, L.; Nichols, J.E.; Guo, J.-T.; et al. Antiviral Activities of ISG20 in Positive-strand RNA Virus Infections. Virology 2011, 409, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, C.R.; Funami, K.; Oshiumi, H.; Mengao, D.; Takaki, H.; Matsumoto, M.; Aly, H.H.; Watashi, K.; Chayama, K.; Seya, T. Interferon-stimulated Gene of 20 kDa Protein (ISG20) Degrades RNA of Hepatitis B Virus to Impede the Replication of HBV in vitro and in vivo. Oncotarget 2016, 7, 68179–68193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, T.Z.D.; Billecocq, A.; Guillemot, L.; Alberts, R.; Gommet, C.; Geffers, R.; Calabrese, K.; Schughart, K.; Bouloy, M.; Montagutelli, X.; et al. A New Mouse Model Reveals a Critical Role for Host Innate Immunity in Resistance to Rift Valley Fever. J. Immunol. 2010, 185, 6146–6156. [Google Scholar] [CrossRef] [PubMed]
- Hinson, E.R.; Joshi, N.S.; Chen, J.H.; Rahner, C.; Jung, Y.W.; Wang, X.; Kaech, S.M.; Cresswell, P. Viperin Is Highly Induced in Neutrophils and Macrophages during Acute and Chronic Lymphocytic Choriomeningitis Virus Infection. J. Immunol. 2010, 184, 5723–5731. [Google Scholar] [CrossRef] [Green Version]
- Cárcamo, J.R.P.; Morell, M.L.; Vázquez, C.A.; Vatansever, S.; Upadhyay, A.S.; Överby, A.K.; Cordo, S.M.; García, C.C. The Interplay between Viperin Antiviral Activity, Lipid Droplets and Junín Mammarenavirus Multiplication. Virology 2018, 514, 216–229. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin Inhibits HIV-1 Release by Directly Tethering Virions to Cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiecki, M.; Omattage, N.S.; Brett, T.J. BST-2/tetherin: Structural Biology, Viral Antagonism, and Immunobiology of a Potent Host Antiviral Factor. Mol. Immunol. 2013, 54, 132–139. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Dong, L.; Chi, X.; Clester, J.C.; Retterer, C.; Spurgers, K.; Kuhn, J.H.; Sandwick, S.; Ruthel, G.; Kota, K.; et al. Infectious Lassa Virus, but Not Filoviruses, Is Restricted by BST-2/Tetherin. J. Virol. 2010, 84, 10569–10580. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, T.; Noda, T.; Urata, S.; Kawaoka, Y.; Yasuda, J. Inhibition of Lassa and Marburg Virus Production by Tetherin. J. Virol. 2008, 83, 2382–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadeh, V.R.; Urata, S.; Sakaguchi, M.; Yasuda, J. Human BST-2/Tetherin Inhibits Junin Virus Release from Host Cells and Its Inhibition is Partially Counteracted by Viral Nucleoprotein. J. Gen. Virol. 2020, 101, 573–586. [Google Scholar] [CrossRef]
- Sakuma, T.; Sakurai, A.; Yasuda, J. Dimerization of Tetherin Is Not Essential for Its Antiviral Activity against Lassa and Marburg Viruses. PLoS ONE 2009, 4, e6934. [Google Scholar] [CrossRef] [Green Version]
- Varela, M.; Piras, I.M.; Mullan, C.; Shi, X.; Tilston-Lunel, N.L.; Pinto, R.M.; Taggart, A.; Welch, S.R.; Neil, S.J.; Kreher, F.; et al. Sensitivity to BST-2 Restriction Correlates with Orthobunyavirus Host Range. Virology 2017, 509, 121–130. [Google Scholar] [CrossRef]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-Stranded RNA Is Produced by Positive-Strand RNA Viruses and DNA Viruses but Not in Detectable Amounts by Negative-Strand RNA Viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [Green Version]
- Mir, M.A.; Panganiban, A.T. The Hantavirus Nucleocapsid Protein Recognizes Specific Features of the Viral RNA Panhandle and Is Altered in Conformation upon RNA Binding. J. Virol. 2005, 79, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Mateer, E.J.; Maruyama, J.; Card, G.E.; Paessler, S.; Huang, C. Lassa Virus, but Not Highly Pathogenic New World Arenaviruses, Restricts Immunostimulatory Double-Stranded RNA Accumulation during Infection. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; Macrae, I.J.; Saphire, E.O. Structure of the Lassa Virus Nucleoprotein Reveals a dsRNA-specific 3’ to 5’ Exonuclease Activity Essential for Immune Suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 2396–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynard, S.; Russier, M.; Fizet, A.; Carnec, X.; Baize, S. Exonuclease Domain of the Lassa Virus Nucleoprotein Is Critical to Avoid RIG-I Signaling and To Inhibit the Innate Immune Response. J. Virol. 2014, 88, 13923–13927. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Huang, Q.; Wang, W.; Dong, H.; Ly, H.; Liang, Y.; Dong, C. Structures of Arenaviral Nucleoproteins with Triphosphate dsRNA Reveal a Unique Mechanism of Immune Suppression. J. Biol. Chem. 2013, 288, 16949–16959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vialat, P.; Billecocq, A.; Kohl, A.; Bouloy, M. The S Segment of Rift Valley Fever Phlebovirus (Bunyaviridae) Carries Determinants for Attenuation and Virulence in Mice. J. Virol. 2000, 74, 1538–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, L.A.; Narayanan, K.; Worthy, M.; Peters, C.J. The S Segment of Punta Toro Virus (Bunyaviridae, Phlebovirus) Is a Major Determinant of Lethality in the Syrian Hamster and Codes for a Type I Interferon Antagonist. J. Virol. 2006, 81, 884–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savellini, G.G.; Weber, F.; Terrosi, C.; Habjan, M.; Martorelli, B.; Cusi, M.G. Toscana Virus Induces Interferon Although Its NSs Protein Reveals Antagonistic Activity. J. Gen. Virol. 2010, 92, 71–79. [Google Scholar] [CrossRef]
- Lihoradova, O.A.; Indran, S.V.; Kalveram, B.; Lokugamage, N.; Head, J.A.; Gong, B.; Tigabu, B.; Juelich, T.L.; Freiberg, A.N.; Ikegami, T. Characterization of Rift Valley Fever Virus MP-12 Strain Encoding NSs of Punta Toro Virus or Sandfly Fever Sicilian Virus. PLoS Negl. Trop. Dis. 2013, 7, e2181. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zheng, B.; Wang, T.; Li, A.; Wan, J.; Qu, J.; Li, C.; Li, D.; Liang, M. NSs Protein of Severe Fever with Thrombocytopenia Syndrome Virus Suppresses Interferon Production through Different Mechanism Than Rift Valley Fever Virus. Acta Virol. 2017, 61, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Kohl, A.; Clayton, R.F.; Weber, F.; Bridgen, A.; Randall, R.E.; Elliott, R.M. Bunyamwera Virus Nonstructural Protein NSs Counteracts Interferon Regulatory Factor 3-Mediated Induction of Early Cell Death. J. Virol. 2003, 77, 7999–8008. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.; Blakqori, G.; Wagner, V.; Banholzer, M.; Kessler, N.; Elliott, R.M.; Haller, O.; Weber, F. Inhibition of RNA Polymerase II Phosphorylation by a Viral Interferon Antagonist. J. Biol. Chem. 2004, 279, 31471–31477. [Google Scholar] [CrossRef] [PubMed]
- Tilston-Lunel, N.L.; Acrani, G.O.; Randall, R.E.; Elliott, R.M. Generation of Recombinant Oropouche Viruses Lacking the Nonstructural Protein NSm or NSs. J. Virol. 2015, 90, 2616–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, G.; Varela, M.; Ratinier, M.; Blomström, A.-L.; Caporale, M.; Seehusen, F.; Hahn, K.; Schnettler, E.; Baumgärtner, W.; Kohl, A.; et al. NSs Protein of Schmallenberg Virus Counteracts the Antiviral Response of the Cell by Inhibiting Its Transcriptional Machinery. J. Gen. Virol. 2014, 95, 1640–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jääskeläinen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala Hantavirus NSs ORFs are Functional and the Products Inhibit Activation of the Interferon-beta Promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Vera-Otarola, J.; Solis, L.; Lowy, F.; Olguín, V.; Angulo, J.; Pino, K.; Tischler, N.D.; Otth, C.; Padula, P.; López-Lastra, M. The Andes Orthohantavirus NSs Protein Antagonizes the Type I Interferon Response by Inhibiting MAVS Signaling. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Ikegami, T.; Narayanan, K.; Won, S.; Kamitani, W.; Peters, C.J.; Makino, S. Dual Functions of Rift Valley Fever Virus NSs Protein: Inhibition of Host mRNA Transcription and Post-transcriptional Downregulation of Protein Kinase PKR. Ann. N. Y. Acad. Sci. 2009, 1171, E75–E85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le May, N.; Dubaele, S.; De Santis, L.P.; Billecocq, A.; Bouloy, M.; Egly, J.-M. TFIIH Transcription Factor, a Target for the Rift Valley Hemorrhagic Fever Virus. Cell 2004, 116, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Cyr, N.; De La Fuente, C.; Lecoq, L.; Guendel, I.; Chabot, P.R.; Kehn-Hall, K.; Omichinski, J.G. A ΩXaV motif in the Rift Valley fever virus NSs protein is essential for degrading p62, forming nuclear filaments and virulence. Proc. Natl. Acad. Sci. USA 2015, 112, 6021–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalveram, B.; Lihoradova, O.; Ikegami, T. NSs Protein of Rift Valley Fever Virus Promotes Posttranslational Downregulation of the TFIIH Subunit p62. J. Virol. 2011, 85, 6234–6243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kainulainen, M.; Habjan, M.; Hubel, P.; Busch, L.; Lau, S.; Colinge, J.; Superti-Furga, G.; Pichlmair, A.; Weber, F. Virulence Factor NSs of Rift Valley Fever Virus Recruits the F-Box Protein FBXO3 To Degrade Subunit p62 of General Transcription Factor TFIIH. J. Virol. 2014, 88, 3464–3473. [Google Scholar] [CrossRef] [Green Version]
- Copeland, A.M.; Van Deusen, N.M.; Schmaljohn, C.S. Rift Valley Fever Virus NSS Gene Expression Correlates with a Defect in Nuclear mRNA Export. Virology 2015, 486, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Le May, N.; Mansuroglu, Z.; Léger, P.; Josse, T.; Blot, G.; Billecocq, A.; Flick, R.; Jacob, Y.; Bonnefoy, E.; Bouloy, M. A SAP30 Complex Inhibits IFN-β Expression in Rift Valley Fever Virus Infected Cells. PLoS Pathog. 2008, 4, e13. [Google Scholar] [CrossRef] [Green Version]
- Rezelj, V.V.; Överby, A.K.; Elliott, R.M. Generation of Mutant Uukuniemi Viruses Lacking the Nonstructural Protein NSs by Reverse Genetics Indicates that NSs Is a Weak Interferon Antagonist. J. Virol. 2015, 89, 4849–4856. [Google Scholar] [CrossRef] [Green Version]
- Simons, J.F.; Hellman, U.; Pettersson, R.F. Uukuniemi Virus S RNA Segment: Ambisense Coding Strategy, Packaging of Complementary Strands into Virions, and Homology to Members of the Genus Phlebovirus. J. Virol. 1990, 64, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikegami, T.; Won, S.; Peters, C.J.; Makino, S. Rift Valley Fever Virus NSs mRNA Is Transcribed from an Incoming Anti-Viral-Sense S RNA Segment. J. Virol. 2005, 79, 12106–12111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, B.; Li, P.; Zhang, S.; Li, A.; Liang, M.; Li, D.; Elliott, R.M. Reverse Genetics System for Severe Fever with Thrombocytopenia Syndrome Virus. J. Virol. 2014, 89, 3026–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbruggen, P.; Ruf, M.; Blakqori, G.; Överby, A.K.; Heidemann, M.; Eick, D.; Weber, F. Interferon Antagonist NSs of La Crosse Virus Triggers a DNA Damage Response-like Degradation of Transcribing RNA Polymerase II. J. Biol. Chem. 2011, 286, 3681–3692. [Google Scholar] [CrossRef] [Green Version]
- Schoen, A.; Lau, S.; Verbruggen, P.; Weber, F. Elongin C Contributes to RNA Polymerase II Degradation by the Interferon Antagonist NSs of La Crosse Orthobunyavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Gouzil, J.; Fablet, A.; Lara, E.; Caignard, G.; Cochet, M.; Kundlacz, C.; Palmarini, M.; Varela, M.; Breard, E.; Sailleau, C.; et al. Nonstructural Protein NSs of Schmallenberg Virus Is Targeted to the Nucleolus and Induces Nucleolar Disorganization. J. Virol. 2017, 91, e01263–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, S.M.; Borg, N.A.; Dixit, V.M. Ubiquitin in the Activation and Attenuation of Innate Antiviral Immunity. J. Exp. Med. 2016, 213, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Sun, S.-C. Ubiquitin Signaling in Immune Responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 Ubiquitin Ligase is Essential for RIG-I-mediated Antiviral Activity. Nat. Cell Biol. 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.T.; Boone, D.L.; et al. De-ubiquitination and Ubiquitin Ligase Domains of A20 Downregulate NF-κB Signalling. Nat. Cell Biol. 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Kayagaki, N.; Phung, Q.; Chan, S.; Chaudhari, R.; Quan, C.; O’Rourke, K.M.; Eby, M.; Pietras, E.; Cheng, G.; Bazan, J.F.; et al. DUBA: A Deubiquitinase That Regulates Type I Interferon Production. Science 2007, 318, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, E.; Martin, S.G.; Grolla, A.; Czub, M.; Feldmann, H.; Flick, R. Sequence Determination of the Crimean–Congo Hemorrhagic Fever Virus L Segment. Virology 2004, 321, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Honig, J.E.; Osborne, J.C.; Nichol, S.T. Crimean–Congo Hemorrhagic Fever Virus Genome L RNA Segment and Encoded Protein. Virology 2004, 321, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, P.J.; Widen, S.G.; Wood, T.G.; Guzman, H.; Tesh, R.B.; Vasilakis, N. A Global Genomic Characterization of Nairoviruses Identifies Nine Discrete Genogroups with Distinctive Structural Characteristics and Host-Vector Associations. Am. J. Trop. Med. Hyg. 2016, 94, 1107–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-M.; Yang, J.; Sun, H.-R.; Xin, X.; Wang, H.-D.; Chen, J.-P.; Adams, M.J. Genomic Analysis of Rice Stripe Virus Zhejiang Isolate Shows the Presence of an OTU-like Domain in the RNA1 Protein and a Novel Sequence Motif Conserved within the Intergenic Regions of Ambisense Segments of Tenuiviruses. Arch. Virol. 2007, 152, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Gu, X.; Li, J.; Liang, C. The N-terminal Cysteine Protease Domain of Rice Stripe Tenuivirus Pc1 Possesses Deubiquitinating Enzyme Activity. Virus Genes 2021, 57, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Frias-Staheli, N.; Giannakopoulos, N.V.; Kikkert, M.; Taylor, S.L.; Bridgen, A.; Paragas, J.; Richt, J.A.; Rowland, R.R.; Schmaljohn, C.S.; Lenschow, D.J.; et al. Ovarian Tumor Domain-Containing Viral Proteases Evade Ubiquitin- and ISG15-Dependent Innate Immune Responses. Cell Host Microbe 2007, 2, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Holzer, B.; Bakshi, S.; Bridgen, A.; Baron, M.D. Inhibition of Interferon Induction and Action by the Nairovirus Nairobi Sheep Disease Virus/Ganjam Virus. PLoS ONE 2011, 6, e28594. [Google Scholar] [CrossRef] [Green Version]
- Bakshi, S.; Holzer, B.; Bridgen, A.; McMullan, G.; Quinn, D.G.; Baron, M.D. Dugbe Virus Ovarian Tumour Domain Interferes with Ubiquitin/ISG15-regulated Innate Immune Cell Signalling. J. Gen. Virol. 2013, 94, 298–307. [Google Scholar] [CrossRef]
- Kocabas, F.; Aslan, G.S. Fluorometric CCHFV OTU Protease Assay with Potent Inhibitors. Virus Genes 2015, 51, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Scholte, F.E.; Zivcec, M.; Dzimianski, J.V.; Deaton, M.K.; Spengler, J.R.; Welch, S.R.; Nichol, S.T.; Pegan, S.D.; Spiropoulou, C.F.; Bergeron, É. Crimean-Congo Hemorrhagic Fever Virus Suppresses Innate Immune Responses via a Ubiquitin and ISG15 Specific Protease. Cell Rep. 2017, 20, 2396–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akutsu, M.; Ye, Y.; Virdee, S.; Chin, J.W.; Komander, D. Molecular Basis for Ubiquitin and ISG15 Cross-reactivity in Viral Ovarian Tumor Domains. Proc. Natl. Acad. Sci. USA 2011, 108, 2228–2233. [Google Scholar] [CrossRef] [Green Version]
- Capodagli, G.C.; McKercher, M.A.; Baker, E.A.; Masters, E.M.; Brunzelle, J.S.; Pegan, S.D. Structural Analysis of a Viral Ovarian Tumor Domain Protease from the Crimean-Congo Hemorrhagic Fever Virus in Complex with Covalently Bonded Ubiquitin. J. Virol. 2011, 85, 3621–3630. [Google Scholar] [CrossRef] [Green Version]
- Van Kasteren, P.; Beugeling, C.; Ninaber, D.; Frias-Staheli, N.; Van Boheemen, S.; García-Sastre, A.; Snijder, E.; Kikkert, M. Arterivirus and Nairovirus Ovarian Tumor Domain-Containing Deubiquitinases Target Activated RIG-I To Control Innate Immune Signaling. J. Virol. 2011, 86, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Capodagli, G.C.; Deaton, M.K.; Baker, E.A.; Lumpkin, R.J.; Pegan, S.D. Diversity of Ubiquitin and ISG15 Specificity among Nairoviruses’ Viral Ovarian Tumor Domain Proteases. J. Virol. 2013, 87, 3815–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaton, M.K.; Dzimianski, J.V.; Daczkowski, C.M.; Whitney, G.K.; Mank, N.J.; Parham, M.M.; Bergeron, E.; Pegan, S.D. Biochemical and Structural Insights into the Preference of Nairoviral DeISGylases for Interferon-Stimulated Gene Product 15 Originating from Certain Species. J. Virol. 2016, 90, 8314–8327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzimianski, J.V.; Beldon, B.S.; Daczkowski, C.M.; Goodwin, O.Y.; Scholte, F.E.M.; Bergeron, É.; Pegan, S.D. Probing the Impact of Nairovirus Genomic Diversity on Viral Ovarian Tumor Domain Protease (vOTU) Structure and Deubiquitinase Activity. PLoS Pathog. 2019, 15, e1007515. [Google Scholar] [CrossRef] [PubMed]
- Devignot, S.; Kromer, T.; Mirazimi, A.; Weber, F. ISG15 Overexpression Compensates the Defect of Crimean-Congo Hemorrhagic Fever Virus Polymerase Bearing a Protease-inactive Ovarian Tumor Domain. PLoS Neglected Trop. Dis. 2020, 14, e0008610. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.L.; Ahrens, P.; Bright, P.M.; Ankel, H. Interferon Induces a 15-kilodalton Protein Exhibiting Marked Homology to Ubiquitin. J. Biol. Chem. 1987, 262, 11315–11323. [Google Scholar] [CrossRef]
- Loeb, K.R.; Haas, A.L. The Interferon-inducible 15-kDa Ubiquitin Homolog Conjugates to Intracellular Proteins. J. Biol. Chem. 1992, 267, 7806–7813. [Google Scholar] [CrossRef]
- Perng, Y.-C.; Lenschow, D.J. ISG15 in Antiviral Immunity and Beyond. Nat. Rev. Genet. 2018, 16, 423–439. [Google Scholar] [CrossRef]
- Freitas, B.T.; Scholte, F.E.; Bergeron, É.; Pegan, S.D. How ISG15 Combats Viral Infection. Virus Res. 2020, 286, 198036. [Google Scholar] [CrossRef]
- Dzimianski, J.V.; Scholte, F.E.M.; Williams, I.L.; Langley, C.; Freitas, B.T.; Spengler, J.R.; Bergeron, É.; Pegan, S.D. Determining the Molecular Drivers of Species-specific Interferon-stimulated Gene Product 15 Interactions with Nairovirus Ovarian Tumor Domain Proteases. PLoS ONE 2019, 14, e0226415. [Google Scholar] [CrossRef] [Green Version]
- Gori-Savellini, G.; Valentini, M.; Cusi, M.G. Toscana Virus NSs Protein Inhibits the Induction of Type I Interferon by Interacting with RIG-I. J. Virol. 2013, 87, 6660–6667. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Briese, T.; Lipkin, W.I. Z Proteins of New World Arenaviruses Bind RIG-I and Interfere with Type I Interferon Induction. J. Virol. 2009, 84, 1785–1791. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Ly, H.; Liang, Y. The Z Proteins of Pathogenic but Not Nonpathogenic Arenaviruses Inhibit RIG-i-Like Receptor-Dependent Interferon Production. J. Virol. 2015, 89, 2944–2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Sobrido, L.; Zúñiga, E.I.; Rosario, D.; García-Sastre, A.; De La Torre, J.C. Inhibition of the Type I Interferon Response by the Nucleoprotein of the Prototypic Arenavirus Lymphocytic Choriomeningitis Virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Sobrido, L.; Giannakas, P.; Cubitt, B.; García-Sastre, A.; De La Torre, J.C. Differential Inhibition of Type I Interferon Induction by Arenavirus Nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, W.W.S.I.; Ortiz-Riaño, E.; Pythoud, C.; Kunz, S.; De La Torre, J.C.; Martínez-Sobrido, L. Arenavirus Nucleoproteins Prevent Activation of Nuclear Factor Kappa B. J. Virol. 2012, 86, 8185–8197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pythoud, C.; Rodrigo, W.W.S.I.; Pasqual, G.; Rothenberger, S.; Martinez-Sobrido, L.; De La Torre, J.C.; Kunz, S. Arenavirus Nucleoprotein Targets Interferon Regulatory Factor-Activating Kinase IKK. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.L.; Krempel, R.L.; Schmaljohn, C.S. Inhibition of TNF-α-induced Activation of NF-κB by Hantavirus Nucleocapsid Proteins. Ann. N. Y. Acad. Sci. 2009, 1171, E86–E93. [Google Scholar] [CrossRef]
- Ontiveros, S.J.; Li, Q.; Jonsson, C.B. Modulation of Apoptosis and Immune Signaling Pathways by the Hantaan Virus Nucleocapsid Protein. Virology 2010, 401, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Bian, G.; Wang, K.; Feng, T.; Dai, J. Effects of Different Doses of Nucleocapsid Protein from Hantaan Virus A9 Strain on Regulation of Interferon Signaling. Viral Immunol. 2015, 28, 448–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.L.; Frias-Staheli, N.; García-Sastre, A.; Schmaljohn, C.S. Hantaan Virus Nucleocapsid Protein Binds to Importin α Proteins and Inhibits Tumor Necrosis Factor Alpha-Induced Activation of Nuclear Factor Kappa B. J. Virol. 2008, 83, 1271–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimica, V.; Dalrymple, N.A.; Roth, E.; Nasonov, A.; Mackow, E.R. An Innate Immunity-Regulating Virulence Determinant Is Uniquely Encoded within the Andes Virus Nucleocapsid Protein. MBio 2014, 5, e01088–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.J.; Gorbunova, E.E.; Mackow, E.R. Unique Interferon Pathway Regulation by the Andes Virus Nucleocapsid Protein Is Conferred by Phosphorylation of Serine 386. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, J.R.; Prescott, J.; Brown, K.S.; Best, S.M.; Ebihara, H.; Feldmann, H. Antagonism of Type I Interferon Responses by New World Hantaviruses. J. Virol. 2010, 84, 11790–11801. [Google Scholar] [CrossRef] [Green Version]
- Alff, P.J.; Gavrilovskaya, I.N.; Gorbunova, E.; Endriss, K.; Chong, Y.; Geimonen, E.; Sen, N.; Reich, N.C.; Mackow, E.R. The Pathogenic NY-1 Hantavirus G1 Cytoplasmic Tail Inhibits RIG-I- and TBK-1-Directed Interferon Responses. J. Virol. 2006, 80, 9676–9686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alff, P.J.; Sen, N.; Gorbunova, E.; Gavrilovskaya, I.N.; Mackow, E.R. The NY-1 Hantavirus Gn Cytoplasmic Tail Coprecipitates TRAF3 and Inhibits Cellular Interferon Responses by Disrupting TBK1-TRAF3 Complex Formation. J. Virol. 2008, 82, 9115–9122. [Google Scholar] [CrossRef] [Green Version]
- Matthys, V.S.; Cimica, V.; Dalrymple, N.A.; Glennon, N.B.; Bianco, C.; Mackow, E.R. Hantavirus GnT Elements Mediate TRAF3 Binding and Inhibit RIG-I/TBK1-Directed Beta Interferon Transcription by Blocking IRF3 Phosphorylation. J. Virol. 2014, 88, 2246–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, B.; Qi, X.; Wu, X.; Liang, M.; Li, C.; Cardona, C.J.; Xu, W.; Tang, F.; Li, Z.; Wu, B.; et al. Suppression of the Interferon and NF-κB Responses by Severe Fever with Thrombocytopenia Syndrome Virus. J. Virol. 2012, 86, 8388–8401. [Google Scholar] [CrossRef] [Green Version]
- Santiago, F.W.; Covaleda, L.M.; Sanchez-Aparicio, M.T.; Silvas, J.A.; Diaz-Vizarreta, A.C.; Patel, J.R.; Popov, V.; Yu, X.-J.; García-Sastre, A.; Aguilar, P.V. Hijacking of RIG-I Signaling Proteins into Virus-Induced Cytoplasmic Structures Correlates with the Inhibition of Type I Interferon Responses. J. Virol. 2014, 88, 4572–4585. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Qi, X.; Qu, B.; Zhang, Z.; Liang, M.; Li, C.; Cardona, C.J.; Li, D.; Xing, Z. Evasion of Antiviral Immunity through Sequestering of TBK1/IKK /IRF3 into Viral Inclusion Bodies. J. Virol. 2013, 88, 3067–3076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, Y.-J.; Wang, M.; Deng, M.; Shen, S.; Liu, W.; Cao, W.-C.; Deng, F.; Wang, Y.-Y.; Hu, Z.; Wang, H. Viral Suppression of Innate Immunity via Spatial Isolation of TBK1/IKKε from Mitochondrial Antiviral Platform. J. Mol. Cell Biol. 2014, 6, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Bai, M.; Qi, X.; Li, C.; Liang, M.; Li, D.; Cardona, C.J.; Xing, Z. Suppression of the IFN-α and -β Induction through Sequestering IRF7 into Viral Inclusion Bodies by Nonstructural Protein NSs in Severe Fever with Thrombocytopenia Syndrome Bunyavirus Infection. J. Immunol. 2019, 202, 841–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, M.; Igarashi, M.; Koshiba, T.; Irie, T.; Takada, A.; Ichinohe, T. Two Conserved Amino Acids within the NSs of Severe Fever with Thrombocytopenia Syndrome Phlebovirus Are Essential for Anti-interferon Activity. J. Virol. 2018, 92, e00706-18. [Google Scholar] [CrossRef] [Green Version]
- Spiropoulou, C.F.; Albariño, C.G.; Ksiazek, T.G.; Rollin, P.E. Andes and Prospect Hill Hantaviruses Differ in Early Induction of Interferon although Both Can Downregulate Interferon Signaling. J. Virol. 2007, 81, 2769–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, Y.-J.; Feng, K.; Min, Y.-Q.; Cao, W.-C.; Wang, M.; Deng, F.; Hu, Z.; Wang, H. Disruption of Type I Interferon Signaling by the Nonstructural Protein of Severe Fever with Thrombocytopenia Syndrome Virus via the Hijacking of STAT2 and STAT1 into Inclusion Bodies. J. Virol. 2015, 89, 4227–4236. [Google Scholar] [CrossRef] [Green Version]
- Kainulainen, M.; Lau, S.; Samuel, C.E.; Hornung, V.; Weber, F. NSs Virulence Factor of Rift Valley Fever Virus Engages the F-Box Proteins FBXW11 and β-TRCP1 To Degrade the Antiviral Protein Kinase PKR. J. Virol. 2016, 90, 6140–6147. [Google Scholar] [CrossRef] [Green Version]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Kota, K.P.; Whitehouse, C.A.; Bavari, S. Protein Kinase R Degradation Is Essential for Rift Valley Fever Virus Infection and Is Regulated by SKP1-CUL1-F-box (SCF)FBXW11-NSs E3 Ligase. PLoS Pathog. 2016, 12, e1005437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalveram, B.; Ikegami, T. Toscana Virus NSs Protein Promotes Degradation of Double-Stranded RNA-Dependent Protein Kinase. J. Virol. 2013, 87, 3710–3718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Mir, M.A. Andes Virus Nucleocapsid Protein Interrupts Protein Kinase R Dimerization to Counteract Host Interference in Viral Protein Synthesis. J. Virol. 2014, 89, 1628–1639. [Google Scholar] [CrossRef] [Green Version]
- Gale, M.; Blakely, C.M.; Kwieciszewski, B.; Tan, S.-L.; Dossett, M.; Tang, N.M.; Korth, M.J.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Control of PKR Protein Kinase by Hepatitis C Virus Nonstructural 5A Protein: Molecular Mechanisms of Kinase Regulation. Mol. Cell. Biol. 1998, 18, 5208–5218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faburay, B.; LaBeaud, A.D.; McVey, D.S.; Wilson, W.C.; Richt, J.A. Current Status of Rift Valley Fever Vaccine Development. Vaccines 2017, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jégouic, S.; Joffret, M.-L.; Blanchard, C.; Riquet, F.B.; Perret, C.; Pelletier, I.; Colbère-Garapin, F.; Rakoto-Andrianarivelo, M.; Delpeyroux, F. Recombination between Polioviruses and Co-Circulating Coxsackie A Viruses: Role in the Emergence of Pathogenic Vaccine-Derived Polioviruses. PLoS Pathog. 2009, 5, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Makoschey, B.; Van Kilsdonk, E.; Hubers, W.R.; Vrijenhoek, M.P.; Smit, M.; Schreur, P.J.W.; Kortekaas, J.; Moulin, V. Rift Valley Fever Vaccine Virus Clone 13 Is Able to Cross the Ovine Placental Barrier Associated with Foetal Infections, Malformations, and Stillbirths. PLoS Negl. Trop. Dis. 2016, 10, e0004550. [Google Scholar] [CrossRef] [PubMed]
- Scholte, F.E.M.; Hua, B.L.; Spengler, J.R.; Dzimianski, J.V.; Coleman-McCray, J.D.; Welch, S.R.; McMullan, L.K.; Nichol, S.T.; Pegan, S.D.; Spiropoulou, C.F.; et al. Stable Occupancy of the Crimean-Congo Hemorrhagic Fever Virus-Encoded Deubiquitinase Blocks Viral Infection. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Genus | Species | Virus (Abbreviation) |
|---|---|---|---|
| Arenaviridae | Mammarenavirus | Argentinian mammarenavirus | Junín virus (JUNV) |
| Brazilian mammarenavirus | Sabiá virus (SBAV) | ||
| Guanarito mammarenavirus | Guanarito virus (GTOV) | ||
| Lassa mammarenavirus | Lassa virus (LASV) | ||
| Lymphocytic choriomeningitis mammarenavirus | Lymphocytic choriomeningitis virus (LCMV) | ||
| Machupo mammarenavirus | Machupo virus (MACV) | ||
| Hantaviridae | Orthohantavirus | Andes orthohantavirus | Andes virus (ANDV) |
| Hantaan orthohantavirus | Hantaan virus (HTNV) | ||
| Prospect Hill orthohantavirus | Prospect Hill virus (PHV) | ||
| Puumala orthohantavirus | Puumala orthohantavirus (PUUV) | ||
| Seoul orthohantavirus | Seoul virus (SEOV) | ||
| Sin Nombre orthohantavirus | New York virus (NYV) | ||
| Sin Nombre virus (SNV) | |||
| Tula orthohantavirus | Tula virus (TULV) | ||
| Nairoviridae | Orthonairovirus | Crimean-Congo hemorrhagic fever orthonairovirus | Crimean-Congo hemorrhagic fever virus (CCHFV) |
| Dugbe orthonairovirus | Dugbe virus (DUGV) | ||
| Nairobi sheep disease orthonairovirus | Nairobi sheep disease virus (NSDV) | ||
| Thiafora orthonairovirus | Erve virus (ERVEV) | ||
| Peribunyaviridae | Orthobunyavirus | Akabane orthobunyavirus | Akabane virus (AKAV) |
| Bunyamwera orthobunyavirus | Bunyamwera virus (BUNV) | ||
| Cache Valley orthobunyavirus | Cache Valley virus (CVV) | ||
| La Crosse orthobunyavirus | La Crosse virus (LACV) | ||
| Oropouche orthobynyavirus | Oropouche virus (OROV) | ||
| Schmallenberg orthobunyavirus | Sathuperi virus (SATV) | ||
| Schmallenberg virus (SBV) | |||
| Phenuiviridae | Banyangvirus | Guertu banyangvirus | Guertu virus (GTV) |
| Heartland banyangvirus | Heartland virus (HRTV) | ||
| Huaiyangshan banyangvirus | Severe fever with thrombocytopenia syndrome virus (SFTSV) | ||
| Phlebovirus | Punta Toro phlebovirus | Punta Toro virus (PTV) | |
| Rift Valley fever phlebovirus | Rift Valley fever virus (RVFV) | ||
| Sandfly fever Naples phlebovirus | Sandfly fever Naples virus (SFNV) | ||
| Toscana virus (TOSV) | |||
| Uukuniemi phlebovirus | Uukuniemi virus (UUKV) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lerolle, S.; Freitas, N.; Cosset, F.-L.; Legros, V. Host Cell Restriction Factors of Bunyaviruses and Viral Countermeasures. Viruses 2021, 13, 784. https://doi.org/10.3390/v13050784
Lerolle S, Freitas N, Cosset F-L, Legros V. Host Cell Restriction Factors of Bunyaviruses and Viral Countermeasures. Viruses. 2021; 13(5):784. https://doi.org/10.3390/v13050784
Chicago/Turabian StyleLerolle, Solène, Natalia Freitas, François-Loïc Cosset, and Vincent Legros. 2021. "Host Cell Restriction Factors of Bunyaviruses and Viral Countermeasures" Viruses 13, no. 5: 784. https://doi.org/10.3390/v13050784
APA StyleLerolle, S., Freitas, N., Cosset, F. -L., & Legros, V. (2021). Host Cell Restriction Factors of Bunyaviruses and Viral Countermeasures. Viruses, 13(5), 784. https://doi.org/10.3390/v13050784