
Diversity and Distribution of Viruses Infecting Wild and Domesticated Phaseolus spp. in the Mesoamerican Center of Domestication

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Library Construction and Small RNA Sequencing
2.3. Small RNA Bioinformatic Analysis
2.4. Validation of Viral Sequences by RT-PCR and Sequencing
2.5. Estimation of Virus Prevalence
2.6. Phylogenetic Analysis
3. Results
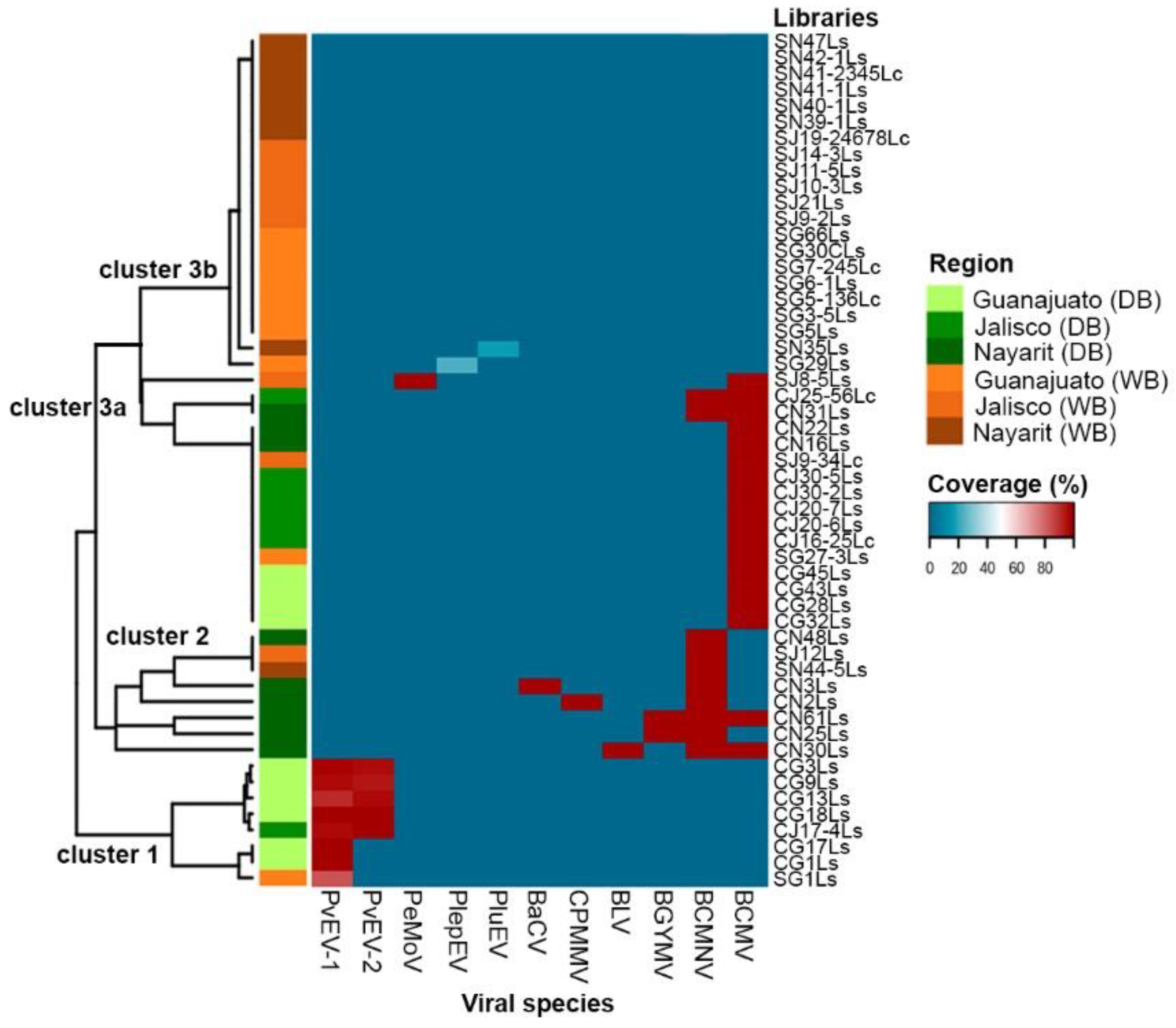
3.1. Viral Species Present in the Domesticated and Wild Bean
3.2. Mixed Infections and Prevalence of Viruses in Domesticated and Wild Bean
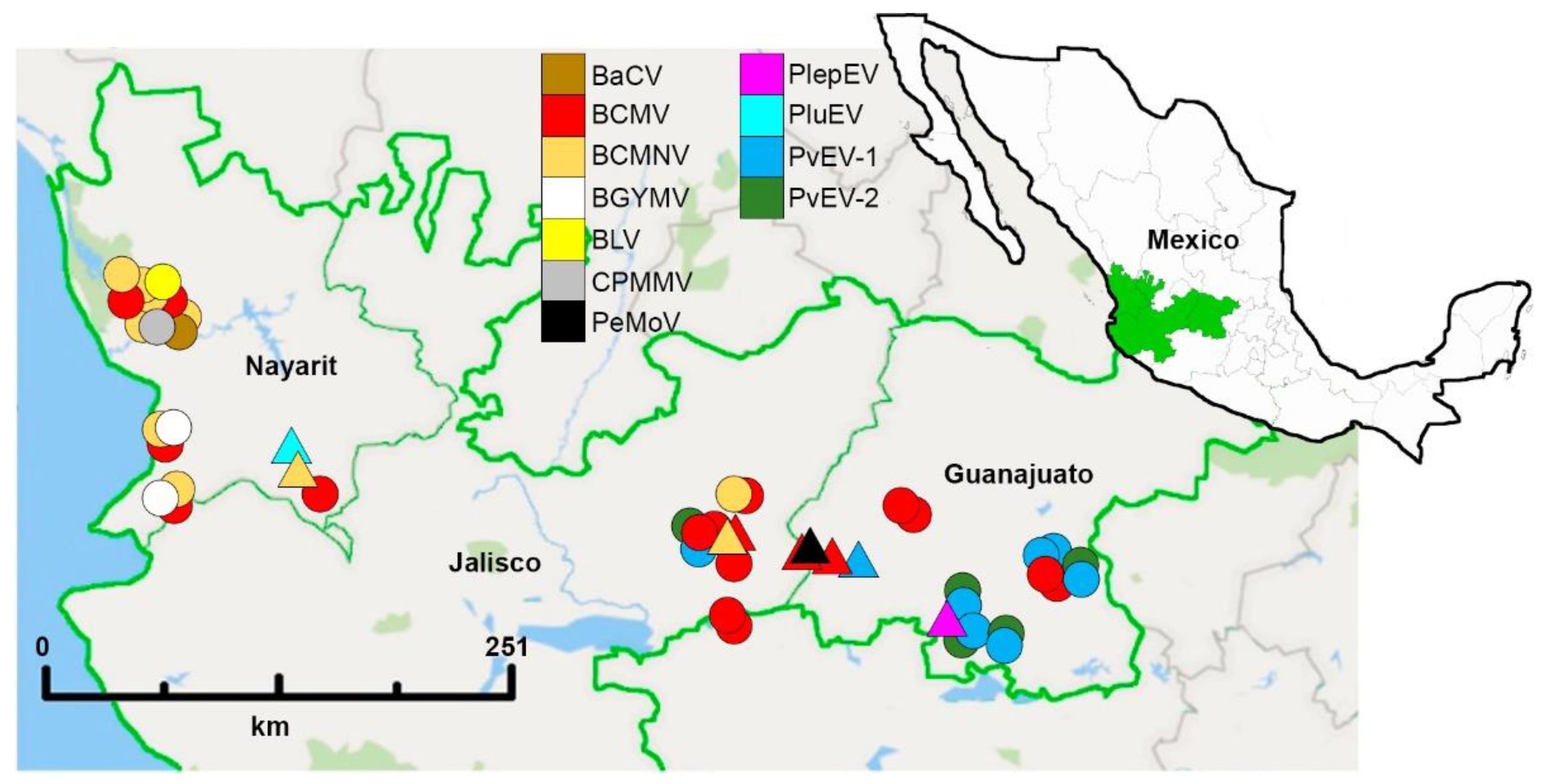
3.3. Viral Distribution Patterns in Domesticated and Wild Bean
3.4. Identification of Novel Viral Species and Viral-Like Sequences
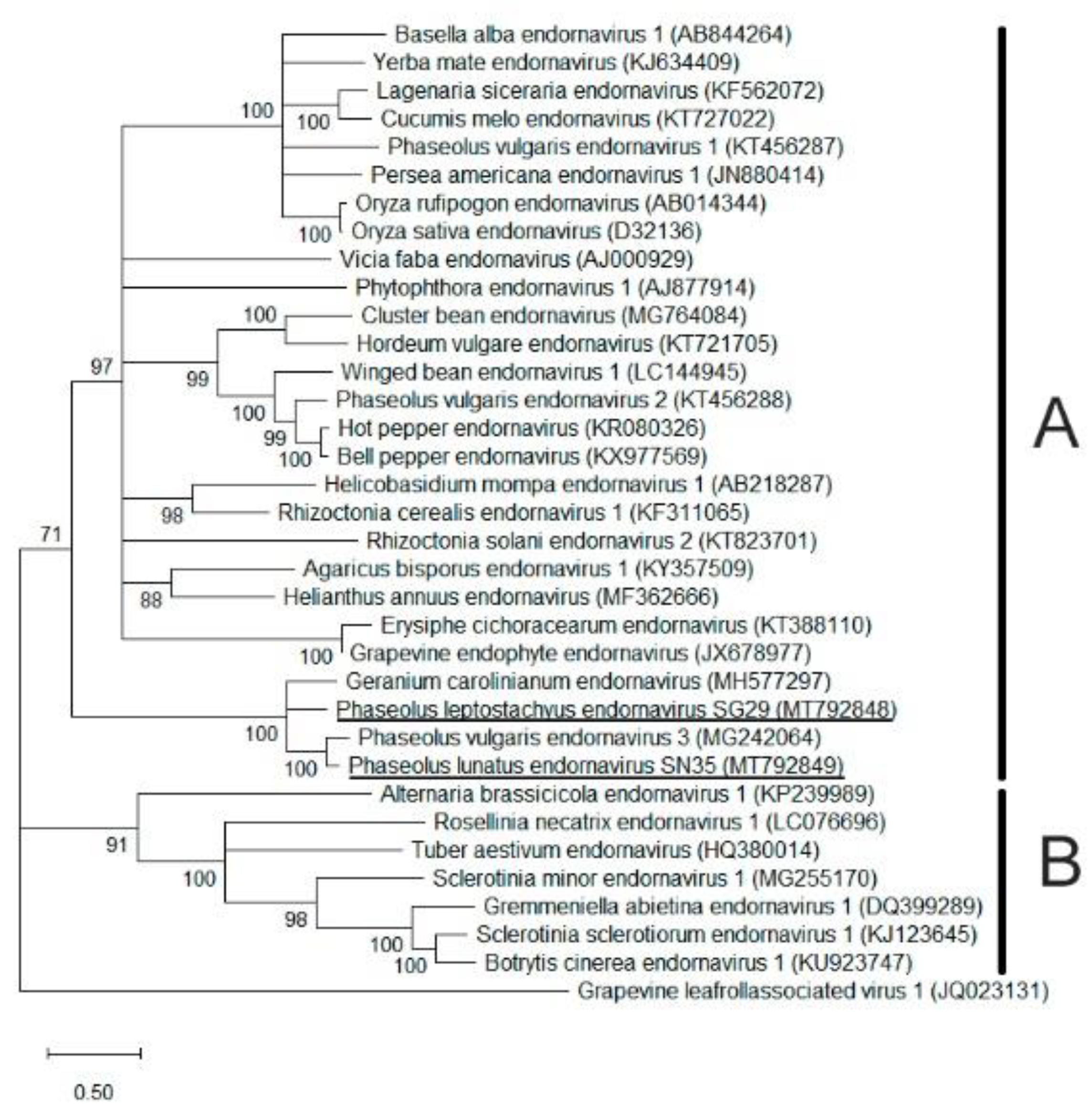
3.4.1. Identification of Two Putative Alphaendornaviruses
3.4.2. Identification of Virus-Like Sequences
4. Discussion
4.1. Identification and Distribution of Viruses in Domesticated and Wild Bean
4.2. Prevalence of Viruses in Domesticated and Wild Bean
4.3. Characteristics of Known Viruses Identified
4.4. Putative Novel Viruses Identified
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, R.A. Plant virus emergence and evolution: Origins, new encounter scenarios, factors driving emergence, effects of changing world conditions, and prospects for control. Virus Res. 2009, 141, 113–130. [Google Scholar] [CrossRef]
- Morales, J.F. Common beans. In Natural Resistance Mechanisms of Plants to Viruses, 1st ed.; Loebenstein, G., Carr, J.P., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 367–382. [Google Scholar]
- FAOSTAT (Food and Agriculture Organization of the United Nations). Available online: http://www.fao.org/faostat/es/#data/QC (accessed on 16 October 2020).
- Bellucci, E.; Bitocchi, E.; Rau, D.; Rodriguez, M.; Biagetti, E.; Giardini, A.; Attene, G.; Nanni, L.; Papa, R. Genomics of origin, domestication and evolution of Phaseolus vulgaris. In Genomics of Plant Genetic Resources, 1st ed.; Tuberosa, R., Graner, A., Frison, E., Eds.; Springer: Dordrecht, The Netherlands, 2014; Volume 1, pp. 483–507. [Google Scholar]
- Rendón-Anaya, M.; Montero-Vargas, J.M.; Saburido-Álvarez, S.; Vlasova, A.; Capella-Gutierrez, S.; Ordaz-Ortiz, J.J.; Aguilar, O.M.; Vianello-Brondani, R.P.; Santalla, M.; Delaye, L.; et al. Genomic history of the origin and domestication of common bean unveils its closest sister species. Genome Biol. 2017, 18, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitocchi, E.; Nanni, L.; Bellucci, E.; Rossi, M.; Giardini, A.; Zeuli, P.S.; Logozzo, G.; Stougaard, J.; McClean, P.; Attene, G.; et al. Mesoamerican origin of the common bean (Phaseolus vulgaris L.) is revealed by sequence data. Proc. Natl. Acad. Sci. USA 2012, 109, E788–E796. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.; Kami, J.A.; Gepts, P. The putative Mesoamerican domestication center of Phaseolus vulgaris is located in the Lerma–Santiago Basin of Mexico. Crop Sci. 2009, 49, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Mwaipopo, B.; Nchimbi-Msolla, S.; Njau, P.J.; Mark, D.; Mbanzibwa, D.R. Comprehensive surveys of Bean common mosaic virus and Bean common mosaic necrosis virus and molecular evidence for occurrence of other Phaseolus vulgaris viruses in Tanzania. Plant Dis. 2018, 102, 2361–2370. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Baizan-Edge, A.; MacFarlane, S.; Torrance, L. Viral diagnostics in plants using next generation sequencing: Computational analysis in practice. Front. Plant Sci. 2017, 8, 1770. [Google Scholar] [CrossRef] [PubMed]
- Kreuze, J. siRNA deep sequencing and assembly: Piecing together viral infections. In Detection and Diagnostics of Plant Pathogens. Plant Pathology in the 21st Century, 1st ed.; Gullino, M.L., Bonants, P.J.M., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 21–38. [Google Scholar]
- Santala, J.; Valkonen, J. Sensitivity of Small RNA-based detection of plant viruses. Front. Microbiol. 2018, 9, 939. [Google Scholar] [CrossRef] [Green Version]
- Chiquito-Almanza, E.; Acosta-Gallegos, J.A.; García-Álvarez, N.C.; Garrido-Ramírez, E.R.; Montero-Tavera, V.; Guevara-Olvera, L.; Anaya-López, J.L. Simultaneous detection of both RNA and DNA viruses infecting dry bean and occurrence of mixed infections by BGYMV, BCMV and BCMNV in the Central-west region of Mexico. Viruses 2017, 9, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.-S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, T.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. Gplots: Various R Programming Tools for Plotting Data. R Package Version 3.1.1. 2020. Available online: https://CRAN.R-project.org/package=gplots (accessed on 17 May 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 17 May 2021).
- Montero-Tavera, V.; Escobedo-Landín, M.A.; Acosta-Gallegos, J.A.; Anaya-López, J.L.; Ruiz-Nieto, J.E. 26S: Novel reference gene from leaves and roots of common bean for biotic stress expression studies based on PCR. Legume Res. 2017, 40, 429–433. [Google Scholar] [CrossRef]
- Sobhy, H.; Colson, P. Gemi: PCR primers prediction from multiple alignments. Comp. Funct. Genom. 2012, 2012, 783138. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Owczarzy, R.; Tataurov, A.V.; Wu, Y.; Manthey, J.A.; McQuisten, K.A.; Almabrazi, H.G.; Pedersen, K.F.; Lin, Y.; Garretson, J.; McEntaggart, N.O.; et al. IDT SciTools: A suite for analysis and design of nucleic acid oligomers. Nucleic Acids Res. 2008, 36, W163–W169. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Marrero, N.; Avalos-Calleros, J.A.; Chiquito-Almanza, E.; Acosta-Gallegos, J.A.; Ambriz-Granados, S.; Anaya-López, J.L.; Argüello-Astorga, G.R. A new begomovirus isolated from a potyvirus-infected bean plant causes asymptomatic infections in bean and N. benthamiana. Arch. Virol. 2020, 165, 1659–1665. [Google Scholar] [CrossRef]
- Alves-Freitas, D.M.; Pinheiro-Lima, B.; Faria, J.C.; Lacorte, C.; Ribeiro, S.G.; Melo, F.L. Double-stranded RNA high-throughput sequencing reveals a new cytorhabdovirus in a bean golden mosaic virus-resistant common bean transgenic line. Viruses 2019, 11, 90. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro-Lima, B.; Pereira-Carvalho, R.C.; Alves-Freitas, D.M.T.; Kitajima, E.W.; Vidal, A.H.; Lacorte, C.; Godinho, M.T.; Fontenele, R.S.; Faria, J.C.; Abreu, E.F.M.; et al. Transmission of the Bean-associated cytorhabdovirus by the whitefly Bemisia tabaci MEAM1. Viruses 2020, 12, 1028. [Google Scholar] [CrossRef]
- Medina-Salguero, A.X.; Cornejo-Franco, J.F.; Grinstead, S.; Mollov, D.; Mowery, J.D.; Flores, F.; Quito-Avila, D.F. Sequencing, genome analysis and prevalence of a cytorhabdovirus discovered in Carica papaya. PLoS ONE 2019, 14, e0215798. [Google Scholar] [CrossRef]
- Okada, R.; Alcalá-Briseño, R.I.; Escalante, C.; Sabanadzovic, S.; Valverde, R.A. Genomic sequence of a novel endornavirus from Phaseolus vulgaris and occurrence in mixed infections with two other endornaviruses. Virus Res. 2018, 257, 63–67. [Google Scholar] [CrossRef]
- Chiquito-Almanza, E.; Acosta-Gallegos, J.A.; García-Álvarez, N.C.; Cuellar, W.; Martínez-Martínez, T.O.; Anaya-López, J.L. Detection of virus damaging the dry bean crop in Nayarit, Mexico. J. Chem. Biol. Physical. Sci. 2014, 4, 48–55. [Google Scholar]
- Narayanasamy, P. Viral and viroid pathogens. In Microbial Plants Pathogens-Detection and Disease Diagnosis; Springer: Dordrecht, The Netherlands, 2011; Volume 3, pp. 12–13. [Google Scholar]
- Voysest, O. Resultados del Primer Vivero Internacional de Rendimiento y Adaptación de Frijol (Phaseolus vulgaris L.) IBYAN 1976. In Frijol Arbustivo; Centro Internacional de Agricultura Tropical (CIAT): Cali, Colombia, 1979; pp. 157–159. [Google Scholar]
- Voysest, O. Vivero Internacional de Rendimiento y Adaptación de Frijol (Phaseolus vulgaris L.) IBYAN 1977. In Frijol de Grano Negro, Frijol de Grano de Diversos Colores; Centro Internacional de Agricultura Tropical (CIAT): Cali, Colombia, 1980; pp. 38–41. [Google Scholar]
- Voysest, O. Vivero Internacional de Rendimiento y Adaptación de Frijol (Phaseolus vulgaris L.) IBYAN 1978. In Frijol Arbustivo con Grano Negro, con Grano de Diversos Colores; Centro Internacional de Agricultura Tropical (CIAT): Cali, Colombia, 1981; pp. 30–47. [Google Scholar]
- Ortiz-Catón, M.; Median-Torres, R.; Valdivia-Bernal, R.; Ortiz-Catón, A.; Alvarado-Casillas, S.; Rodríguez-Blanco, J.R. Mosquitas blancas plaga primaria de hortalizas en Nayarit. Rev. Fuente 2010, 2, 31–40. [Google Scholar]
- Fraile, A.; McLeish, M.J.; Pagán, I.; González-Jara, P.; Piñero, D.; García-Arenal, F. Environmental heterogeneity and the evolution of plant-virus interactions: Viruses in wild pepper populations. Virus Res. 2017, 241, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Sonnante, G.; Stockton, T.; Nodari, R.O.; Becerra Velásquez, V.L.; Gepts, P. Evolution of genetic diversity during the domestication of common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 1994, 89, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, P.; Charles-Dominique, T.; Barakat, M.; Ortet, P.; Fernandez, E.; Filloux, D.; Hartnady, P.; Rebelo, T.A.; Cousins, S.R.; Mesleard, F.; et al. Geometagenomics illuminates the impact of agriculture on the distribution and prevalence of plant viruses at the ecosystem scale. ISME J. 2018, 12, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Chiquito-Almanza, E.; Caballero-Pérez, J.; Guevara-Olvera, L.; Acosta-García, G.; Pérez-Pérez, M.C.I.; Acosta-Gallegos, J.A.; Anaya-López, J.L. First report of Cowpea mild mottle virus infecting cultivated and wild Phaseolus in the Central-Western region of Mexico. Plant Dis. 2018, 102, 1047. [Google Scholar] [CrossRef]
- Jones, R.A.; Salam, M.U.; Maling, T.J.; Diggle, A.J.; Thackray, D.J. Principles of predicting plant virus disease epidemics. Annu. Rev. Phytopathol. 2010, 48, 179–203. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.R.; Gilbertson, R.L. Emerging plant viruses: A diversity of mechanisms and opportunities. In Plant Virus Evolution, 1st ed.; Roossinck, M.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 27–51. [Google Scholar]
- Elena, S.F.; Fraile, A.; García-Arenal, F. Evolution and emergence of plant viruses. Adv. Virus Res. 2014, 88, 161–191. [Google Scholar] [CrossRef] [Green Version]
- Morales, F.J.; Anderson, P.K. The emergence and dissemination of whitefly-transmitted geminiviruses in Latin America. Arch. Virol. 2001, 146, 415–441. [Google Scholar] [CrossRef]
- Freitas-Astua, J.; Dietzgen, R.G.; Walker, P.J.; Blasdell, K.R.; Breyta, R.; Fooks, A.R. Create Twelve New Species in the Genus Cytorhabdovirus, Family Rhabdoviridae. Approved ICTV Proposal. 2019. Available online: https://ictv.global/ICTV/proposals/2019.030M.zip (accessed on 15 October 2020).
- Valverde, R.A.; Khalifa, M.E.; Okada, R.; Fukuhara, T.; Sabanadzovic, S. ICTV virus taxonomy profile: Endornaviridae. J. Gen. Virol. 2019, 100, 1204–1205. [Google Scholar] [CrossRef]
- Fukuhara, T. Endornaviruses: Persistent dsRNA viruses with symbiotic properties in diverse eukaryotes. Virus Genes 2019, 55, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Okada, R.; Yong, C.K.; Valverde, R.A.; Sabanadzovic, S.; Aoki, N.; Hotate, S.; Kiyota, E.; Moriyama, H.; Fukuhara, T. Molecular characterization of two evolutionally distinct endornaviruses coinfecting common bean (Phaseolus vulgaris). J. Gen. Virol. 2013, 94, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Khankhum, S.; Valverde, R.A. Physiological traits of endornavirus-infected and endornavirus-free common bean (Phaseolus vulgaris) cv Black Turtle Soup. Arch. Virol. 2018, 163, 1051–1056. [Google Scholar] [CrossRef]
- Morales, F.L.; Castaño, M. Enfermedades Virales del Frijol Común en América Latina; Centro Internacional de Agricultura Tropical (CIAT): Cali, Colombia, 2008; pp. 9–44. [Google Scholar]
- Morales, F.J.; Castaño, M. Seed transmission characteristics of selected bean common mosaic virus strains in differential bean cultivars. Plant Dis. 1987, 71, 51–53. [Google Scholar] [CrossRef]
- Boland, G.J.; Melzer, M.S.; Hopkin, A.; Higgins, V.; Nassuth, A. Climate change and plant diseases in Ontario. Can. J. Plant Pathol. 2004, 26, 335–350. [Google Scholar] [CrossRef]
- Beebe, S.; Ramirez, J.; Jarvis, A.; Rao, E.M.; Mosquera, G.; Bueno, J.M.; Blair, M.W. Genetic improvement of common beans and the challenges of climate change. In Crop Adaptation to Climate Change; Yadav, S.S., Redden, R., Hatfield, J.L., Lotze-Campen, H., Hall, A.J.W., Eds.; Wiley-Blackwell: Oxford, UK, 2011; pp. 356–369. [Google Scholar]
- Sastry, K.S. Seed-Borne Plant Virus Diseases; Springer: New Delhi, India, 2013; p. 88. [Google Scholar]
- Morales, F.J.; Bos, L. Bean Common Mosaic Virus. Descriptions of Plant Viruses, 337. Association of Applied Biologists, UK. 1998. Available online: http://www.dpvweb.net/dpv/showadpv.php?dpvno=337 (accessed on 28 May 2020).
- Sastry, K.S. Plant Virus and Viroid Diseases in the Tropics: Introduction of Plant Viruses and Sub-Viral Agents, Classification, Assessment of Loss, Transmission and Diagnosis; Springer: Dordrecht, The Netherlands, 2013; p. 110. [Google Scholar]
- Lamas, N.S.; Matos, V.O.R.L.; Alves-Freitas, D.M.T.; Melo, F.L.; Costa, A.F.; Faria, J.C.; Ribeiro, S.G. Occurrence of Cowpea mild mottle virus in common bean and associated weeds in Northeastern Brazil. Plant Dis. 2017, 101, 1828. [Google Scholar] [CrossRef]
- Zanardo, L.G.; Carvalho, C.M. Cowpea mild mottle virus (Carlavirus, Betaflexiviridae): A review. Trop. Plant Pathol. 2017, 42, 417–430. [Google Scholar] [CrossRef]
- Marubayashi, J.M.; Yuki, V.A.; Wutke, E.B. Transmission of Cowpea mild mottle virus by Bemisia tabaci biotype B from plants of beans and soybean. Summa Phytopathol. 2010, 36, 158–160. [Google Scholar] [CrossRef] [Green Version]
- Zanardo, L.G.; Silva, F.N.; Bicalho, A.A.C.; Urquiza, G.P.C.; Lima, A.T.M.; Almeida, A.M.R.; Zerbini, F.M.; Carvalho, C.M. Molecular and biological characterization of Cowpea mild mottle virus isolates infecting soybean in Brazil and evidence of recombination. Plant Pathol. 2014, 63, 456–465. [Google Scholar] [CrossRef]
- European and Mediterranean Plant Protection Organization (EPPO). Cowpea Mild Motte Virus. Categorization. Available online: https://gd.eppo.int/taxon/CPMMV0/categorization (accessed on 10 August 2020).
- Brown, J.K.; Verle-Rodrigues, J.C. Recovery Plan. Cowpea Mild Mottle Virus. 2014. Available online: https://www.ars.usda.gov/ARSUserFiles/00000000/opmp/CPMMV%20Recovery%20Plan.pdf (accessed on 10 August 2020).
- Khankhum, S.; Valverde, R.A.; Pastor-Corrales, M.; Osorno, J.M.; Sabanadzovic, S. Two endornaviruses show differential infection patterns between gene pools of Phaseolus vulgaris. Arch. Virol. 2015, 160, 1131–1137. [Google Scholar] [CrossRef]
- Staginnus, C.; Richert-Poggeler, K.R. Endogenous pararetroviruses: Two-faced travelers in the plant genome. Trends Plant Sci. 2006, 11, 485–491. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Name | Acronym | Genus | Family | Genome * | MCD | Nay. | Genomes Assembled § | Transmission | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Gto. | Jal. | Seed | Vectors | |||||||
| Phaseolus vulgaris alphaendornavirus 1 | PvEV-1 | Alphaendornavirus | Endornaviridae | dsRNA | + | + | 8 | + | U | |
| Phaseolus vulgaris alphaendornavirus 2 | PvEV-2 | Alphaendornavirus | Endornaviridae | dsRNA | + | + | 5 | + | U | |
| Bean common mosaic virus | BCMV | Potyvirus | Potyviridae | ssRNA (+) | + | + | + | 18 | + | Aphids |
| Bean common mosaic necrosis virus | BCMNV | Potyvirus | Potyviridae | ssRNA (+) | + | + | 10 | + | Aphids | |
| Peanut mottle virus | PeMoV | Potyvirus | Potyviridae | ssRNA (+) | + | 1 | + | Aphids | ||
| Cowpea mild mottle virus | CPMMV | Carlavirus | Betaflexiviridae | ssRNA (+) | + | 1 | + | B. tabaci | ||
| Bean-associated cytorhabdovirus | BaCV | Cytorhabdovirus | Rhabdoviridae | ssRNA (-) | + | 1 | U | B. tabaci | ||
| Bean golden yellow mosaic virus DNA A | BGYMV DNA A | Begomovirus | Geminiviridae | ssDNA | + | 2 | − | B. tabaci | ||
| Bean golden yellow mosaic virus DNA B | BGYMV DNA B | Begomovirus | Geminiviridae | ssDNA | + | 2 | − | B. tabaci | ||
| Bean latent virus DNA A | BLV DNA A | Begomovirus | Geminiviridae | ssDNA | + | 1 | U | U | ||
| Bean latent virus DNA B | BLV DNA B | Begomovirus | Geminiviridae | ssDNA | + | 1 | U | U | ||
| Phaseolus leptostachyus alphaendornavirus | PlepEV | Putative Alphaendornavirus ¶ | Endornaviridae | dsRNA | + | U | U | |||
| Phaseolus lunatus alphaendornavirus | PluEV | Putative Alphaendornavirus ¶ | Endornaviridae | dsRNA | + | U | U | |||
| Badnavirus-like sequence | PvBV-CG17 | Putative Badnavirus ¥ | Caulimoviridae | dsDNA | + | U | U | |||
| Badnavirus-like sequence | PcBV-SJ10-3 | Putative Badnavirus ¥ | Caulimoviridae | dsDNA | + | U | U | |||
| Badnavirus-like sequence | PluBV-SN35 | Putative Badnavirus ¥ | Caulimoviridae | dsDNA | + | U | U | |||
| Pararetrovirus-like sequence | PcPRV-SJ10-3 | Not assigned ¥ | Caulimoviridae | dsDNA | + | U | U | |||
| Pararetrovirus-like sequence | PluPRV-SN35 | Not assigned ¥ | Caulimoviridae | dsDNA | + | U | U | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiquito-Almanza, E.; Caballero-Pérez, J.; Acosta-Gallegos, J.A.; Montero-Tavera, V.; Mariscal-Amaro, L.A.; Anaya-López, J.L. Diversity and Distribution of Viruses Infecting Wild and Domesticated Phaseolus spp. in the Mesoamerican Center of Domestication. Viruses 2021, 13, 1153. https://doi.org/10.3390/v13061153
Chiquito-Almanza E, Caballero-Pérez J, Acosta-Gallegos JA, Montero-Tavera V, Mariscal-Amaro LA, Anaya-López JL. Diversity and Distribution of Viruses Infecting Wild and Domesticated Phaseolus spp. in the Mesoamerican Center of Domestication. Viruses. 2021; 13(6):1153. https://doi.org/10.3390/v13061153
Chicago/Turabian StyleChiquito-Almanza, Elizabeth, Juan Caballero-Pérez, Jorge A. Acosta-Gallegos, Victor Montero-Tavera, Luis Antonio Mariscal-Amaro, and José Luis Anaya-López. 2021. "Diversity and Distribution of Viruses Infecting Wild and Domesticated Phaseolus spp. in the Mesoamerican Center of Domestication" Viruses 13, no. 6: 1153. https://doi.org/10.3390/v13061153
APA StyleChiquito-Almanza, E., Caballero-Pérez, J., Acosta-Gallegos, J. A., Montero-Tavera, V., Mariscal-Amaro, L. A., & Anaya-López, J. L. (2021). Diversity and Distribution of Viruses Infecting Wild and Domesticated Phaseolus spp. in the Mesoamerican Center of Domestication. Viruses, 13(6), 1153. https://doi.org/10.3390/v13061153