
Comprehensive Evolutionary Analysis of Complete Epstein–Barr Virus Genomes from Argentina and Other Geographies

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Ethics Statement
2.2. Patients and Samples
2.3. DNA Extraction
2.4. Viral Load Assays
2.5. Library Preparation and Illumina Sequencing
2.6. Publicly Available Sequence Data
2.7. Bioinformatic Data Processing
2.8. Multiple Sequence Alignment and Evolutionary Tests
2.9. Estimation of Evolution Rate
2.10. Gene Ontology-Based Clustering
2.11. Statistical Analysis
3. Results
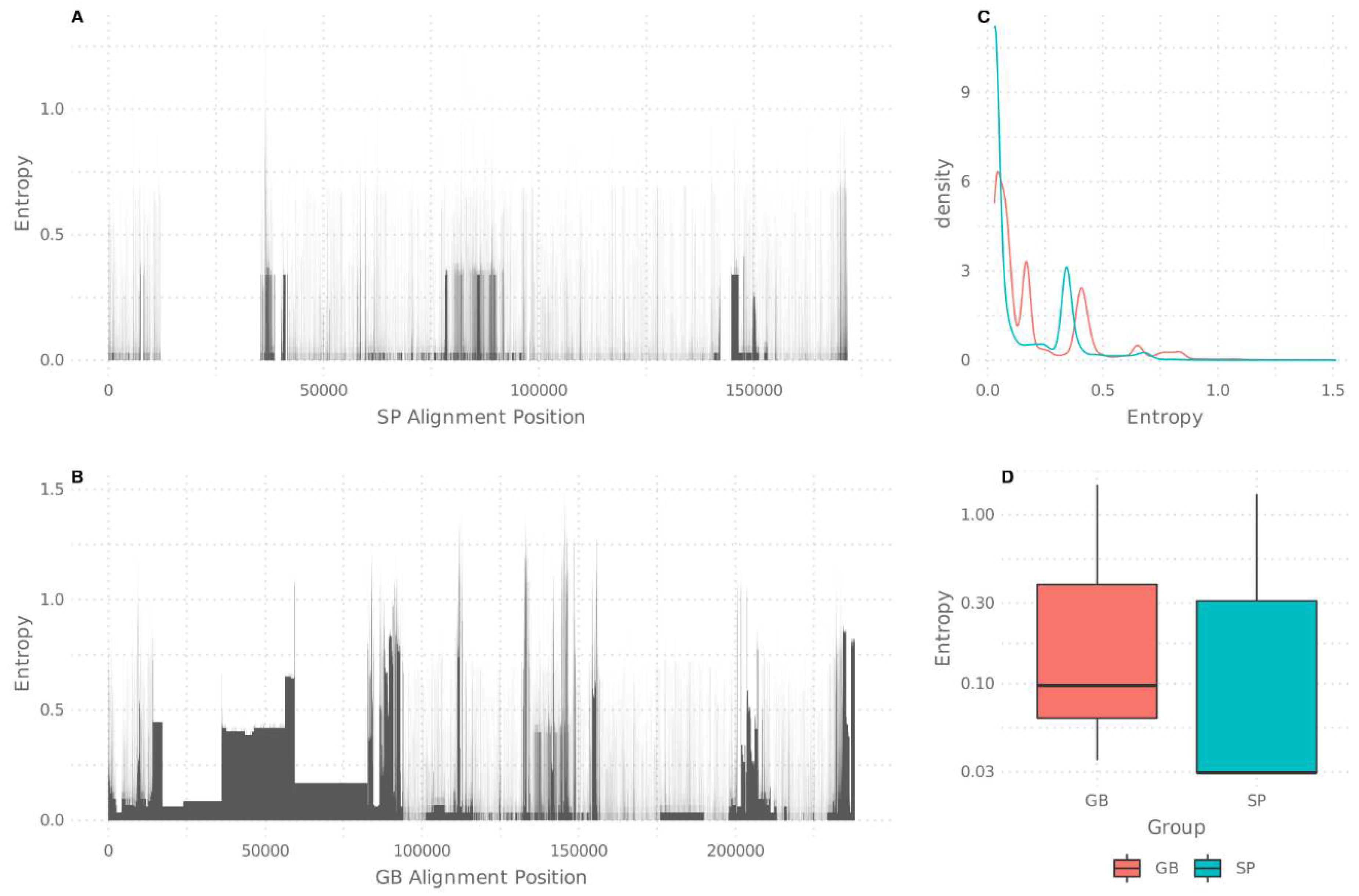
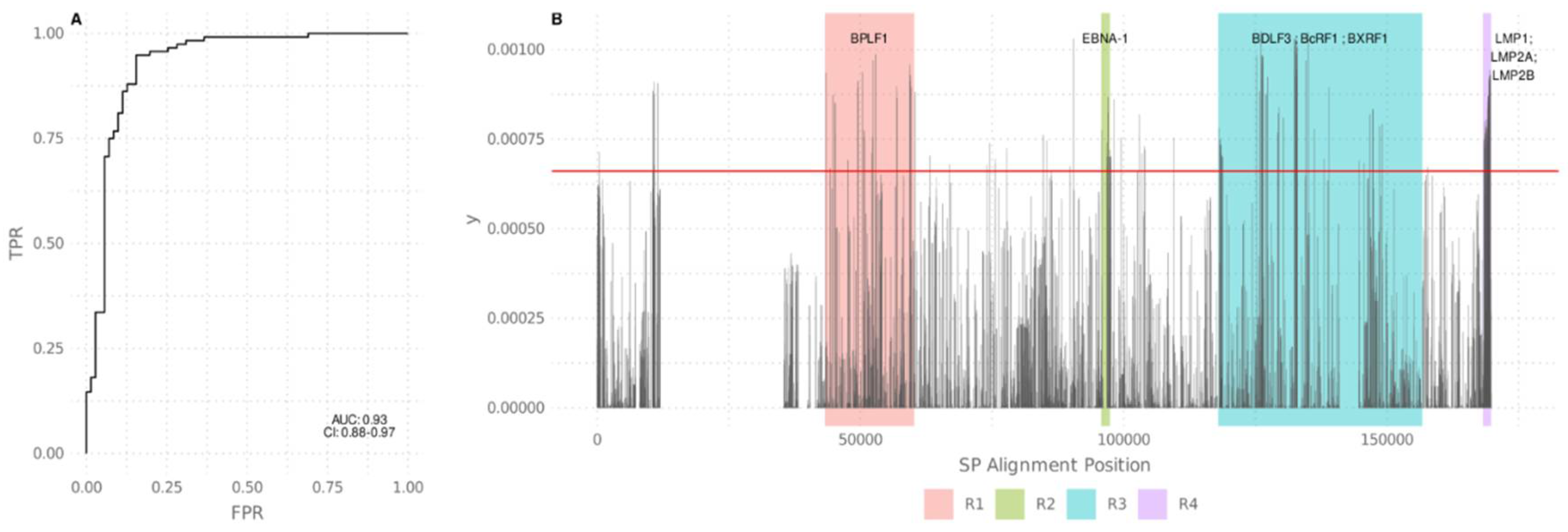
3.1. A Call for a Unified Raw Data Sequence Analysis
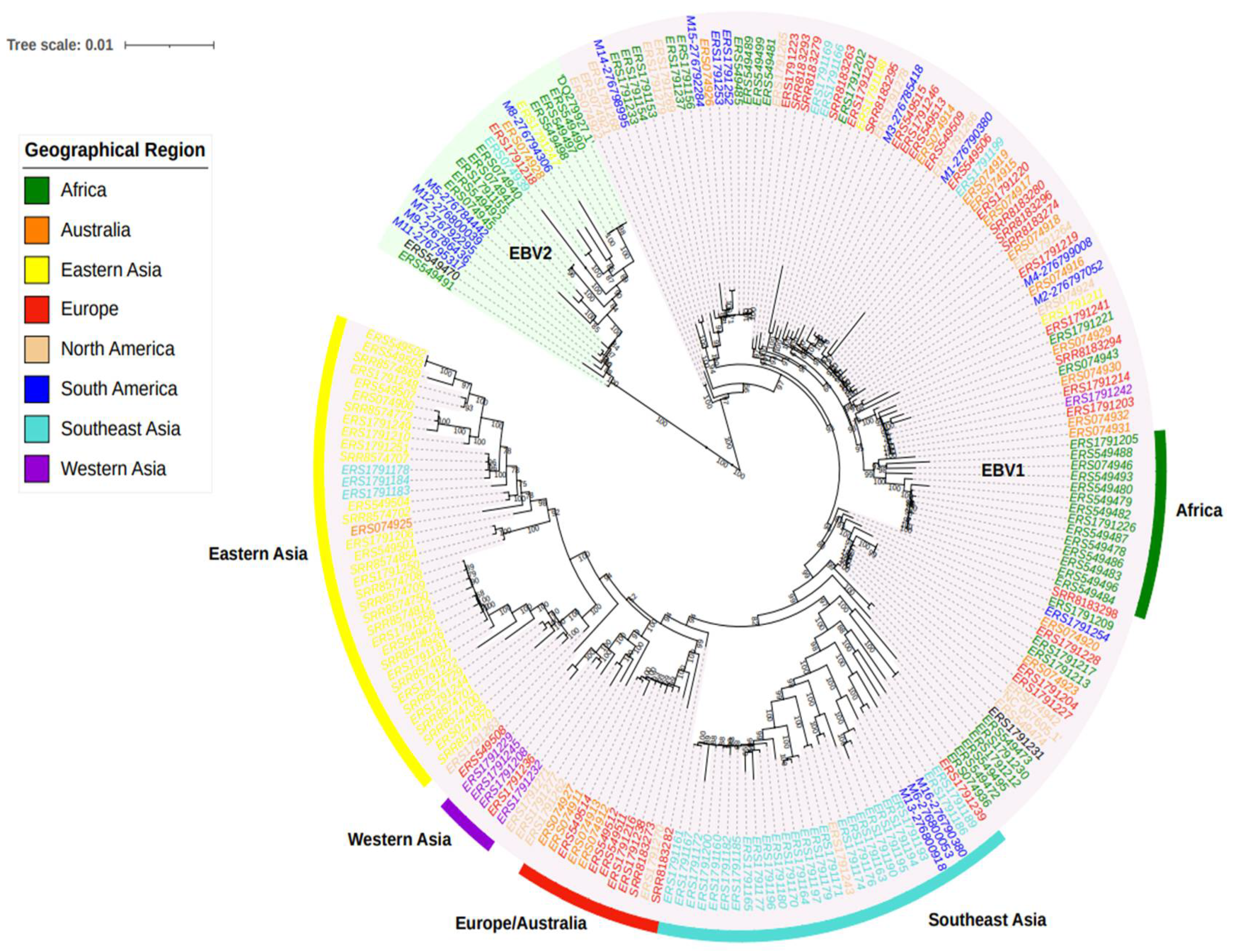
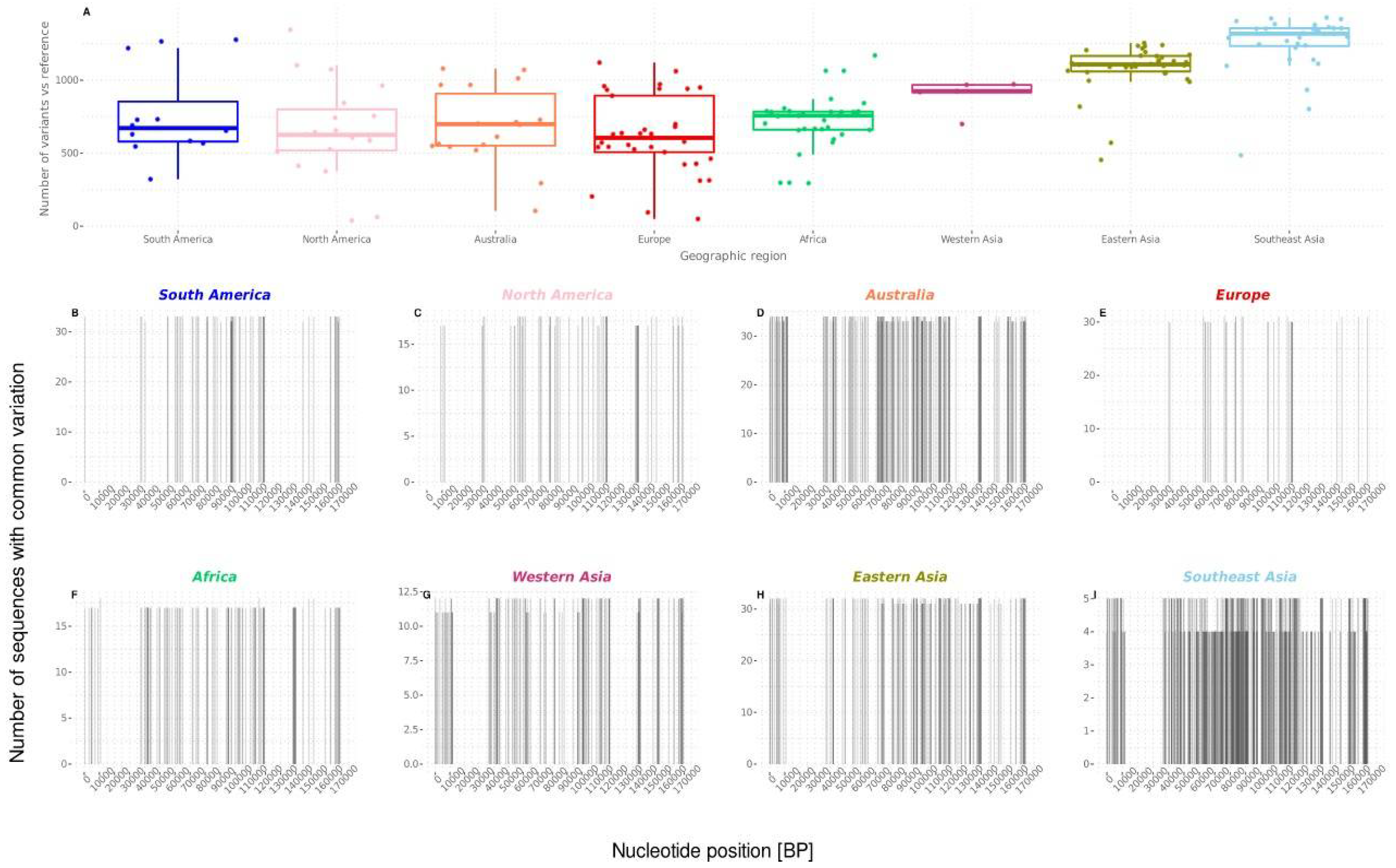
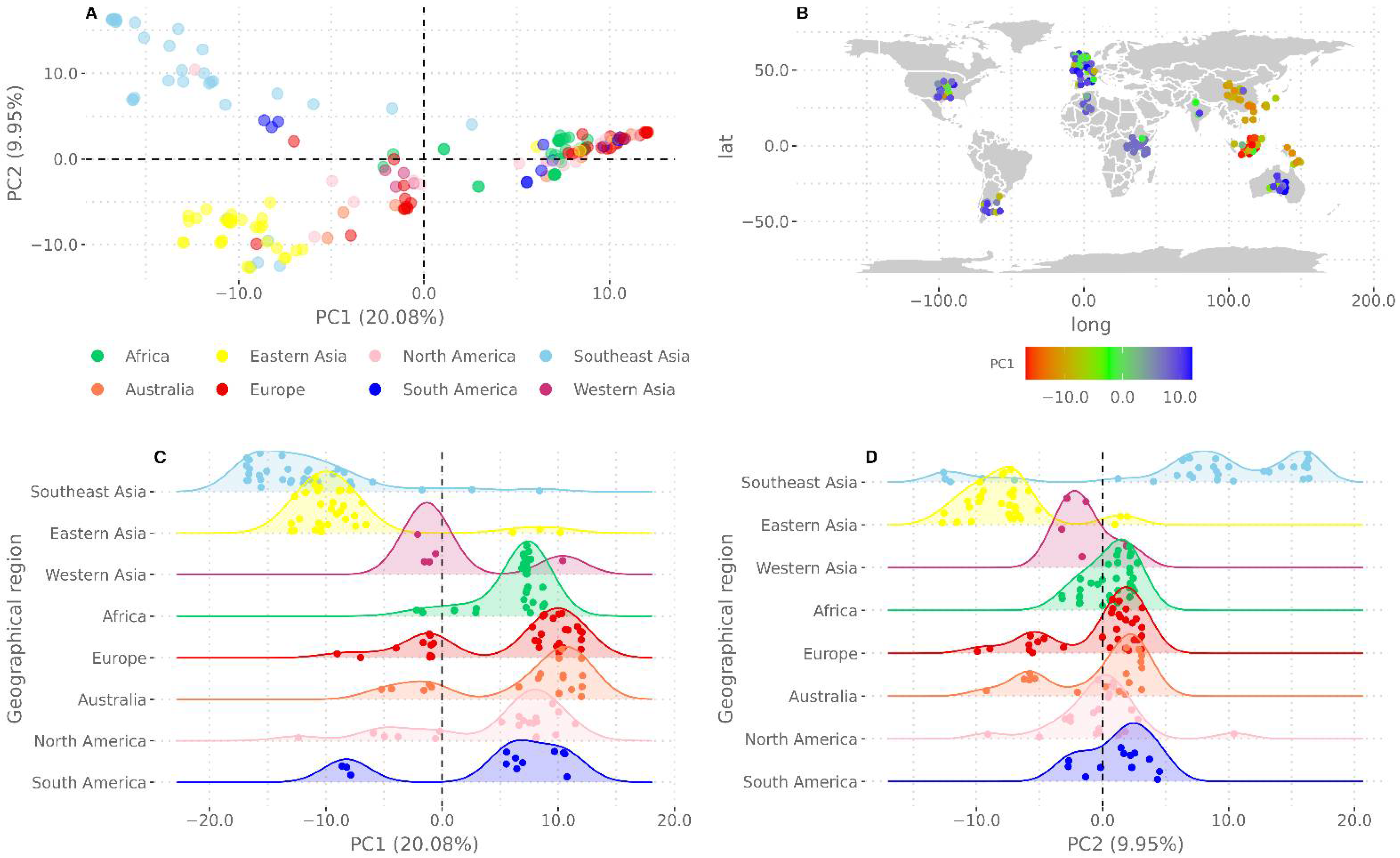
3.2. Phylogenetic Relations, Evolution, and Geographic Segregation
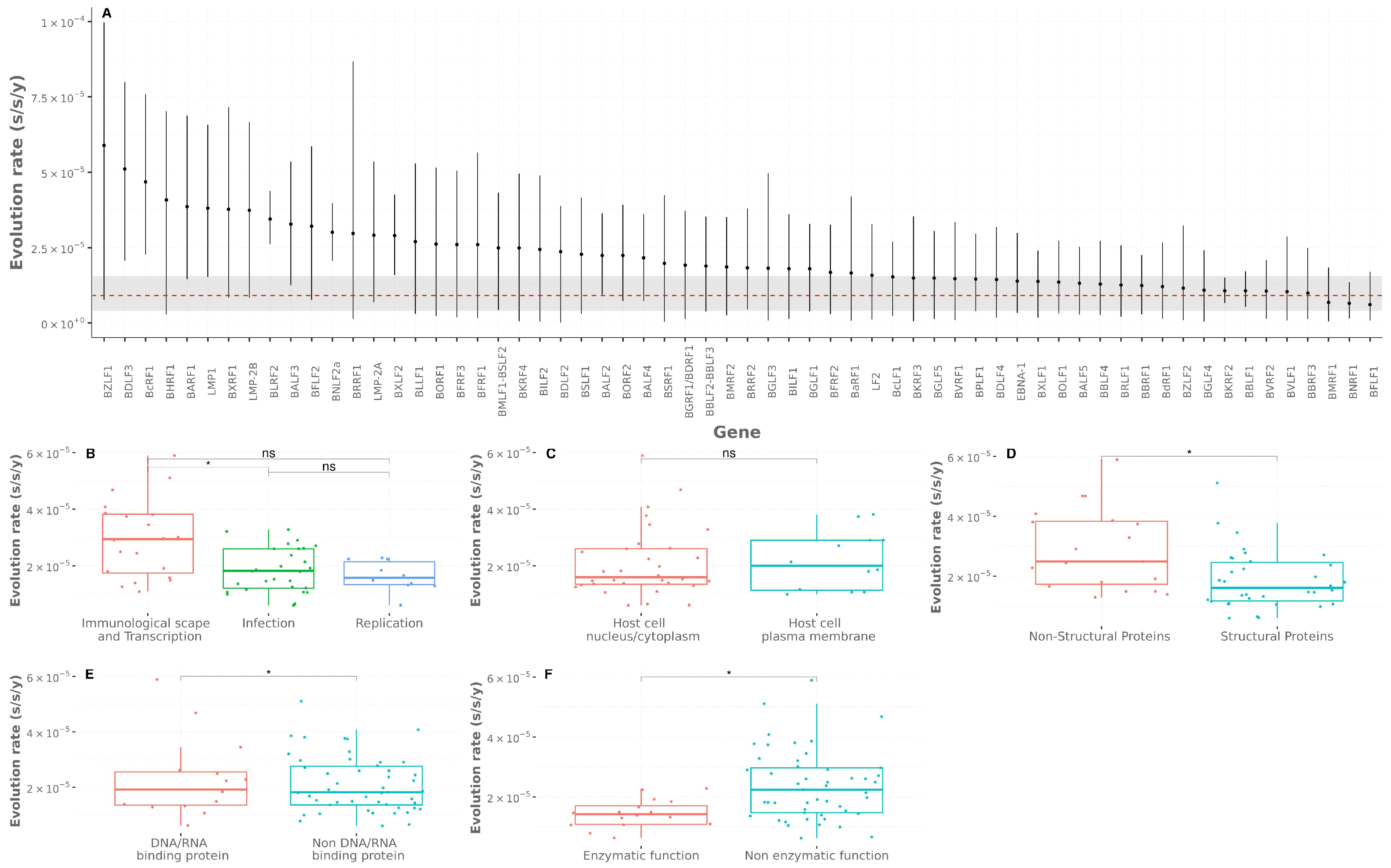
3.3. Genomic Evolutionary Rates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdullah, M.M.B.; Palermo, R.D.; Palser, A.L.; Grayson, N.E.; Kellam, P.; Correia, S.; Szymula, A.; White, R.E. Heterogeneity of the Epstein-Barr Virus (EBV) Major Internal Repeat Reveals Evolutionary Mechanisms of EBV and a Functional Defect in the Prototype EBV Strain B95-8. J. Virol. 2017, 91, e00920-17. [Google Scholar] [CrossRef] [Green Version]
- Luzuriaga, K.; Sullivan, J.L. Infectious Mononucleosis. N. Engl. J. Med. 2010, 362, 1993–2000. [Google Scholar] [CrossRef] [Green Version]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yao, Y.; Chen, H.; Zhang, S.; Cao, S.-M.; Zhang, Z.; Luo, B.; Liu, Z.; Li, Z.; Xiang, T.; et al. Genome sequencing analysis identifies Epstein–Barr virus subtypes associated with high risk of nasopharyngeal carcinoma. Nat. Genet. 2019, 51, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Chan, T.F.; Yang, W.; Shen, J.J.; Lam, K.P.; Kwok, H.; Sham, P.C.; Tsao, S.W.; Kwong, D.L.; Lung, M.L.; et al. High risk Epstein-Barr virus variants characterized by distinct polymorphisms in the EBER locus are strongly associated with nasopharyngeal carcinoma. Int. J. Cancer 2019, 144, 3031–3042. [Google Scholar] [CrossRef] [PubMed]
- Telford, M.; Hughes, D.; Juan, D.; Stoneking, M.; Navarro, A.; Santpere, G. Expanding the Geographic Characterisation of Epstein–Barr Virus Variation through Gene-Based Approaches. Microorganisms 2020, 8, 1686. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetti, M.A.; Gantuz, M.; Altcheh, J.; De Matteo, E.; Chabay, P.A.; Preciado, M.V. Epstein–Barr virus BZLF1 gene polymorphisms: Malignancy related or geographically distributed variants? Clin. Microbiol. Infect. 2014, 20, O861–O869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neves, M.; Marinho-Dias, J.; Ribeiro, J.; Sousa, H. Epstein-Barr virus strains and variations: Geographic or disease-specific variants? J. Med. Virol. 2017, 89, 373–387. [Google Scholar] [CrossRef]
- Chang, C.M.; Yu, K.J.; Mbulaiteye, S.M.; Hildesheim, A.; Bhatia, K. The extent of genetic diversity of Epstein-Barr virus and its geographic and disease patterns: A need for reappraisal. Virus Res. 2009, 143, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Fellner, M.D.; Durand, K.A.; Solernou, V.; Bosaleh, A.; Balbarrey, Z.; de Dávila, M.T.G.; Rodríguez, M.; Irazu, L.; Alonio, L.V.; Picconi, M.A. Epstein–Barr virus load in transplant patients: Early detection of post-transplant lymphoproliferative disorders. Rev. Argent. Microbiol. 2016, 48, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Depledge, D.P.; Palser, A.L.; Watson, S.J.; Lai, I.Y.-C.; Gray, E.; Grant, P.; Kanda, R.K.; Leproust, E.; Kellam, P.; Breuer, J. Specific Capture and Whole-Genome Sequencing of Viruses from Clinical Samples. PLoS ONE 2011, 6, e27805. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazutaka, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- R Development Core Team. The R Reference Manual: Base Package. Network Theory Ltd. 2003. Available online: https://cran.r-project.org/ (accessed on 15 July 2020).
- Thibaut, J. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Gantuz, M.; Lorenzetti, M.A.; Chabay, P.A.; Preciado, M.V. A Novel Recombinant Variant of Latent Membrane Protein 1 from Epstein Barr Virus in Argentina Denotes Phylogeographical Association. PLoS ONE 2017, 12, e0174221. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A.; Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Lawson, R.G.; Jurs, P.C. New index for clustering tendency and its application to chemical problems. J. Chem. Inf. Comput. Sci. 1990, 30, 36–41. [Google Scholar] [CrossRef]
- Kassambara, A. Practical Guide to Cluster Analysis in R: Unsupervised Machine Learning; STHDA, 2017. Available online: http://www.sthda.com/french/ (accessed on 15 July 2020).
- Correia, S.; Bridges, R.; Wegner, F.; Venturini, C.; Palser, A.; Middeldorp, J.; Cohen, J.I.; Lorenzetti, M.A.; Bassano, I.; White, R.; et al. Sequence Variation of Epstein-Barr Virus: Viral Types, Geography, Codon Usage, and Diseases. J. Virol. 2018, 92, e01132-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, F.; Lassalle, F.; Depledge, D.P.; Balloux, F.; Breuer, J. Coevolution of Sites under Immune Selection Shapes Epstein–Barr Virus Population Structure. Mol. Biol. Evol. 2019, 36, 2512–2521. [Google Scholar] [CrossRef]
- Bridges, R.; Correia, S.; Wegner, F.; Venturini, C.; Palser, A.; White, R.E.; Kellam, P.; Breuer, J.; Farrell, P.J. Essential role of inverted repeat in Epstein–Barr virus IR-1 in B cell transformation; geographical variation of the viral genome. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmartin, M. Colonialism/Imperialism. In Key Concepts in Political Geography; SAGE Publications, Inc.: Thousand Oaks, CA, USA, 2014. [Google Scholar] [CrossRef]
- Avena, S.; Via, M.; Ziv, E.; Pérez-Stable, E.J.; Gignoux, C.R.; Dejean, C.; Huntsman, S.; Torres-Mejía, G.; Dutil, J.; Matta, J.L.; et al. Heterogeneity in Genetic Admixture across Different Regions of Argentina. PLoS ONE 2012, 7, e34695. [Google Scholar] [CrossRef] [Green Version]
- Zanella, L.; Riquelme, I.; Buchegger, K.; Abanto, M.; Ili, C.; Brebi, P. A reliable Epstein-Barr Virus classification based on phylogenomic and population analyses. Sci. Rep. 2019, 9, 9829. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Fang, Q.; Zuo, J.; Minhas, V.; Wood, C.; Zhenqiu, L.; Zhang, T. Was Kaposi’s sarcoma-associated herpesvirus introduced into China via the ancient Silk Road? An evolutionary perspective. Arch. Virol. 2017, 162, 3061–3068. [Google Scholar] [CrossRef]
- Kolb, A.W.; Ané, C.; Brandt, C.R.; Kolb, A.W.; Ané, C.; Brandt, C.R. Using HSV-1 Genome Phylogenetics to Track Past Human Migrations. PLoS ONE 2013, 8, e76267. [Google Scholar] [CrossRef] [Green Version]
- Firth, C.; Kitchen, A.; Shapiro, B.; Suchard, M.A.; Holmes, E.C.; Rambaut, A. Using Time-Structured Data to Estimate Evolutionary Rates of Double-Stranded DNA Viruses. Mol. Biol. Evol. 2010, 27, 2038–2051. [Google Scholar] [CrossRef] [Green Version]
- Tzellos, S.; Farrell, P.J. Epstein-Barr Virus Sequence Variation—Biology and Disease. Pathogens 2012, 1, 156–174. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Xu, M.; Liang, L.; Zhang, H.; Xu, R.; Feng, Q.; Feng, L.; Luo, B.; Zeng, Y.-X. Genome-wide analysis of Epstein-Barr virus identifies variants and genes associated with gastric carcinoma and population structure. Tumor Biol. 2017, 39, 1010428317714195. [Google Scholar] [CrossRef] [Green Version]
- Correia, S.; Palser, A.; Karstegl, C.E.; Middeldorp, J.; Ramayanti, O.; Cohen, J.I.; Hildesheim, A.; Fellner, M.D.; Wiels, J.; White, R.; et al. Natural Variation of Epstein-Barr Virus Genes, Proteins, and Primary MicroRNA. J. Virol. 2017, 91, e00375-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzetti, M.A.; Altcheh, J.; Moroni, S.; Moscatelli, G.; Chabay, P.A.; Preciado, M.V. EBNA1 Sequences in Argentinean Pediatric Acute and Latent Epstein-Barr Virus Infection Reflect Circulation of Novel South American Variants. J. Med. Virol. 2010, 82, 1730–1738. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. Evolution of the mutation rate. Trends Genet. 2010, 26, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Ho, S.Y. Molecular clocks. Curr. Biol. 2016, 26, R399–R402. [Google Scholar] [CrossRef] [Green Version]
- Scally, A. The mutation rate in human evolution and demographic inference. Curr. Opin. Genet. Dev. 2016, 41, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Process |
| Viral immunological escape and transcription LMP1, LMP2A, LMP2B, BGLF4, BZLF1, EBNA1, BMLF1-BSLF2, BILF1, BLRF2, BNLF2a, BGLF5, LF2, BCRF1, BHRF1, BRLF1, BcRF1, BARF1, BLDF3, BILF2, BRRF1 |
| Viral infective BBRF3, BGLF3, BZLF2, BcLF1, BXLF2, BSRF1, BDLF1, BdRF1, BFLF1, BPLF1, BFRF3, BDLF2, BLLF1, BFRF1, BFLF2, BKRF2M BVRF1, BMRF2, BGLF1, BBRF2, BALF3, BGRF1/BDRF1, BALF4, BBRF1, BOLF1, BNRF1, BORF1, BVRF2, BBLF1 |
| Viral replicative BMRF1, BSLF1, BBLF2, BALF2, BALF5, BXLF1, BaRF1, BORF2, BBLF4, BKRF3 |
| Cellular Component |
| Host cell plasma membrane BLLF1, BALF4, BXLF2, BKRF2, BMRF2, BBRF3, BBLF1, BZLF2, BILF1, LMP1, LMP2a, LMP2b |
| Host cell nucleus/cytoplasm BMRF1, BGLF4M BPLF1, BFLF1, BLRF2, BXRF1, BGLF1, BBRF2, BSRF1, BDLF1, BORF1, BFRF3, BVRF1, BcLF1, BVRF2, BNRF1, BOLF1, BXLF1, BALF2M BBRF1, EBNA1, BZLF1, BHRF1, BcRF1, BSLFS/BMLF1, BALF5, BGLF5, BBLF4, BALF3, BKRF3, BSLF1, BaRF1 |
| Extracellular space BCRF1, BARF1 |
| Viral Component |
| Structural proteins BLLF1, BALF4, BXLF2, BKRF2, BMRF2, BBRF3, BBLF1, BZLF2, BDLF3, BDLF2, BFRF2, BKRF4, BdRF1, BGLF4, BMRF1, BPLF1, BFLF1, BLRF2, BXRF1, BGLF1, BBRF2, BSRF1, BDLF1, BORF1, BFRF3, BVRF1, BcLF1, BVRF2, BNRF1, BOLF1, BXLF1, BALF2, BBRF1, BALF5 |
| Non structural proteins LMP1, LMP2a, LMP2b, BILF1, BILF2, EBNA1, BZLF1, BcRF1, BCRF1, BARF1, BHRF1, BSLF2-BMLF1, BGLF5, BBLF4, BKRF3, BSLF1, BaRF1 |
| Enzymatic Function |
| Enzymatic function BGLF5, BGLF4, BGRF1/BDRF1, BPLF1, BKRF2, BVRF2, BSLF1, BORF2, BBLF2, BaRF1, BKRF3, BXLF1, BALF5, BBLF4, BLLF3, BMRF1 |
| Non enzymatic function BZLF1, BDLF3, BcRF1, BHRF1, BARF1, LMP1, LMP2a, LMP2b, BLRF2, BNLF2a, BRRF1, BMLF1-BSLF2, BILF2, BGLF3, BILF1, BFRF2, BDLF4, EBNA1, BRLF1, BVLF1, BXRF1, BALF3, BFLF2, BXLF2, BDLF1, BLLF1, BORF1, BFRF1, BFRF3, BKRF4, BDLF2, BALF4, BSRF1, BMRF2, BBRF2, BGLF1, BcLF1, BVRF1, BOLF1, BBRF1, BdRF1, BZLF2, BBLF1, BBRF3, BNRF1, BFLF1, BALF2 |
| DNA Binding Protein |
| DNA/RNA binding protein BGLF5, BGRF1/BDRF1, BKRF2, BSLF1, BBLF2M BALF5, BBLF4, BMRF1, BZLF1, BcRF1, BLRF1, BMLF1-BSRLF2, EBNA1, BORF1, BALF2 |
| Non DNA/RNA binding protein BDLF3, BHRF1, BARF1, LMP1, LMP2a, LMP2b, BNLF2a, BRRF1, BILF2, BGLF3, BILF1, BFRF2, BDLF4, BRLF1, BVLF1, BXRF1, BALF3, BFLF2, BXLF2, BDLF1, BLLF1, BFRF1, BFRF3, BKRF4, BDLF2, BALF4, BSRF1, BMRF2, BBRF2, BGLF1, BcLF1, BVRF1, BOLF1, BBRF1, BdRF1, BZLF2, BBLF1, BBRF3, BNRF1, BFLF1, BGLF4, BPLF1, BVRF2, BORF2, BaRF1, BKRF3, BXLF1, BLLF3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blazquez, A.C.; Berenstein, A.J.; Torres, C.; Izquierdo, A.; Lezama, C.; Moscatelli, G.; De Matteo, E.N.; Lorenzetti, M.A.; Preciado, M.V. Comprehensive Evolutionary Analysis of Complete Epstein–Barr Virus Genomes from Argentina and Other Geographies. Viruses 2021, 13, 1172. https://doi.org/10.3390/v13061172
Blazquez AC, Berenstein AJ, Torres C, Izquierdo A, Lezama C, Moscatelli G, De Matteo EN, Lorenzetti MA, Preciado MV. Comprehensive Evolutionary Analysis of Complete Epstein–Barr Virus Genomes from Argentina and Other Geographies. Viruses. 2021; 13(6):1172. https://doi.org/10.3390/v13061172
Chicago/Turabian StyleBlazquez, Ana Catalina, Ariel José Berenstein, Carolina Torres, Agustín Izquierdo, Carol Lezama, Guillermo Moscatelli, Elena Noemí De Matteo, Mario Alejandro Lorenzetti, and María Victoria Preciado. 2021. "Comprehensive Evolutionary Analysis of Complete Epstein–Barr Virus Genomes from Argentina and Other Geographies" Viruses 13, no. 6: 1172. https://doi.org/10.3390/v13061172
APA StyleBlazquez, A. C., Berenstein, A. J., Torres, C., Izquierdo, A., Lezama, C., Moscatelli, G., De Matteo, E. N., Lorenzetti, M. A., & Preciado, M. V. (2021). Comprehensive Evolutionary Analysis of Complete Epstein–Barr Virus Genomes from Argentina and Other Geographies. Viruses, 13(6), 1172. https://doi.org/10.3390/v13061172