
Bacteriophages fEV-1 and fD1 Infect Yersinia pestis

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Phages, and Media
2.2. Phage Titration, Host Screening, and Efficiency of Plating
2.3. Phage Production and Purification
2.4. Electron Microscopy
2.5. Phage Genome Extraction, Sequencing, and Analysis
2.6. PCR and Sanger Sequencing
2.7. Restriction Endonuclease Analysis
2.8. Sample Preparation for Mass Spectrometry
2.9. Sample Preparation for Mass Spectrometry
2.10. Mass Spectrometry Data Analysis
2.11. Phage Growth Curves
3. Results and Discussion
3.1. Electron Microscopy
3.2. Phage Host Specificity and Efficiency of Plating
3.3. Phage Growth Curves
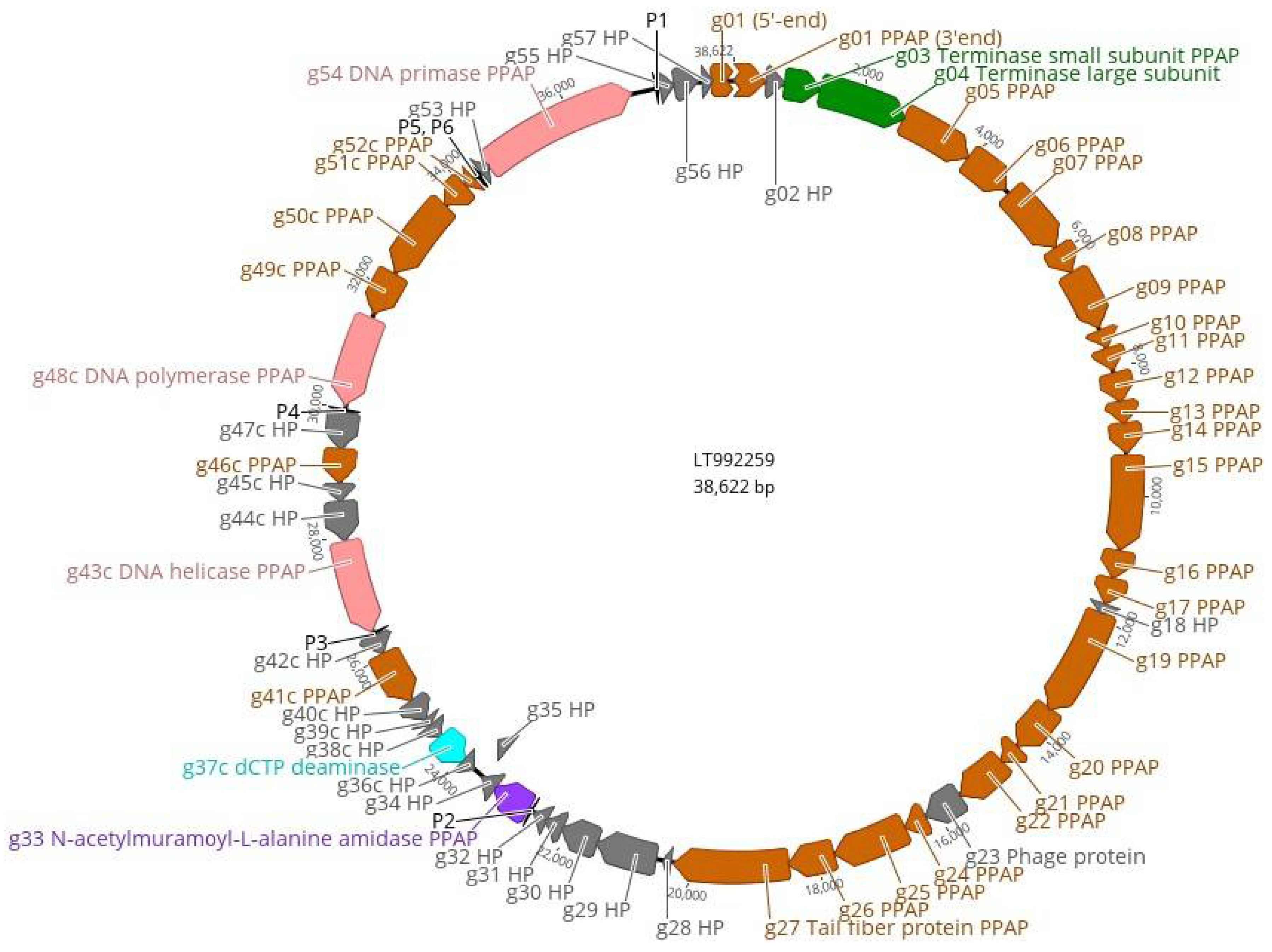
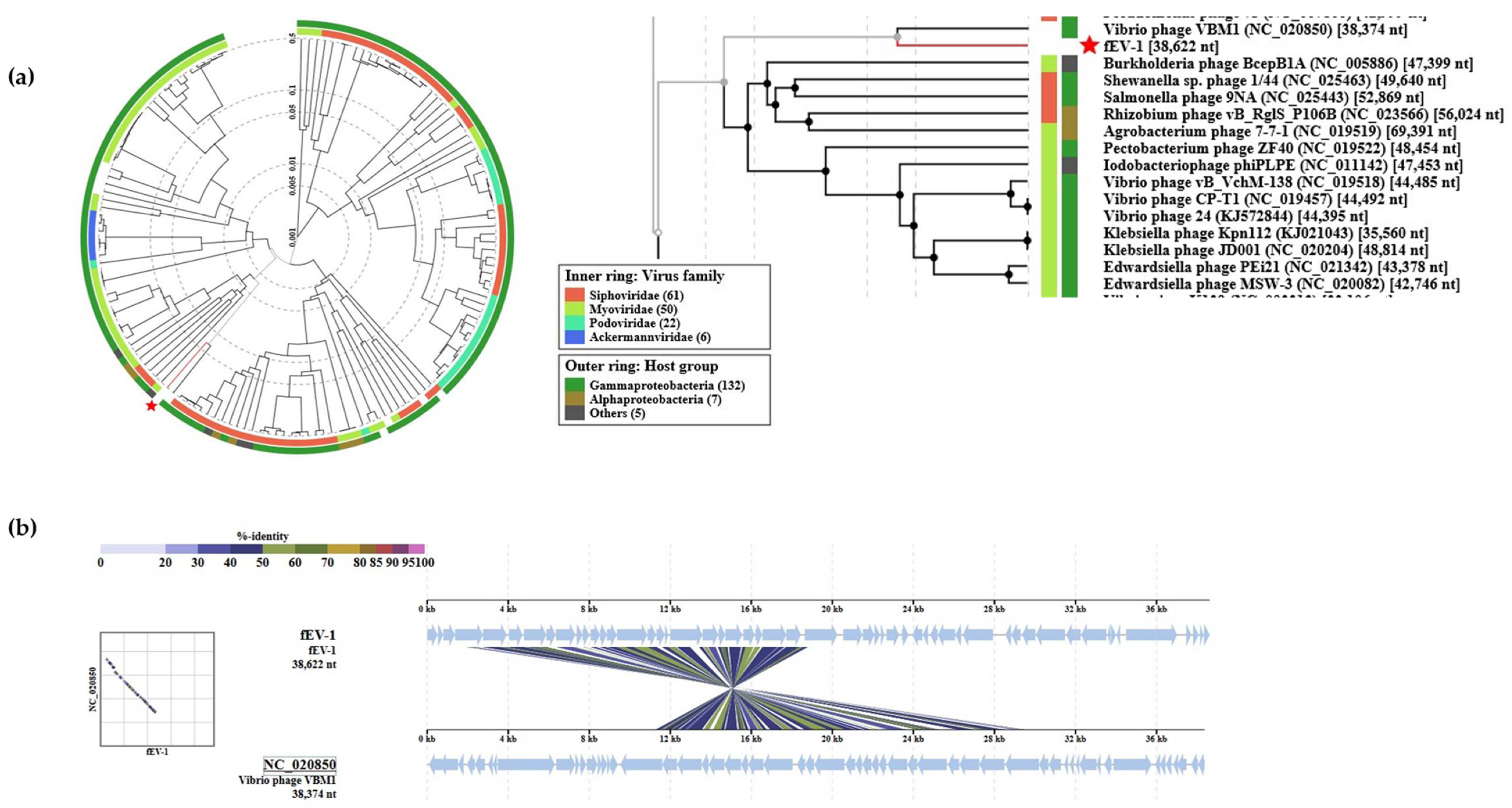
3.4. The Genome and Taxonomic Position of fEV-1
3.5. The Proteome of fEV-1
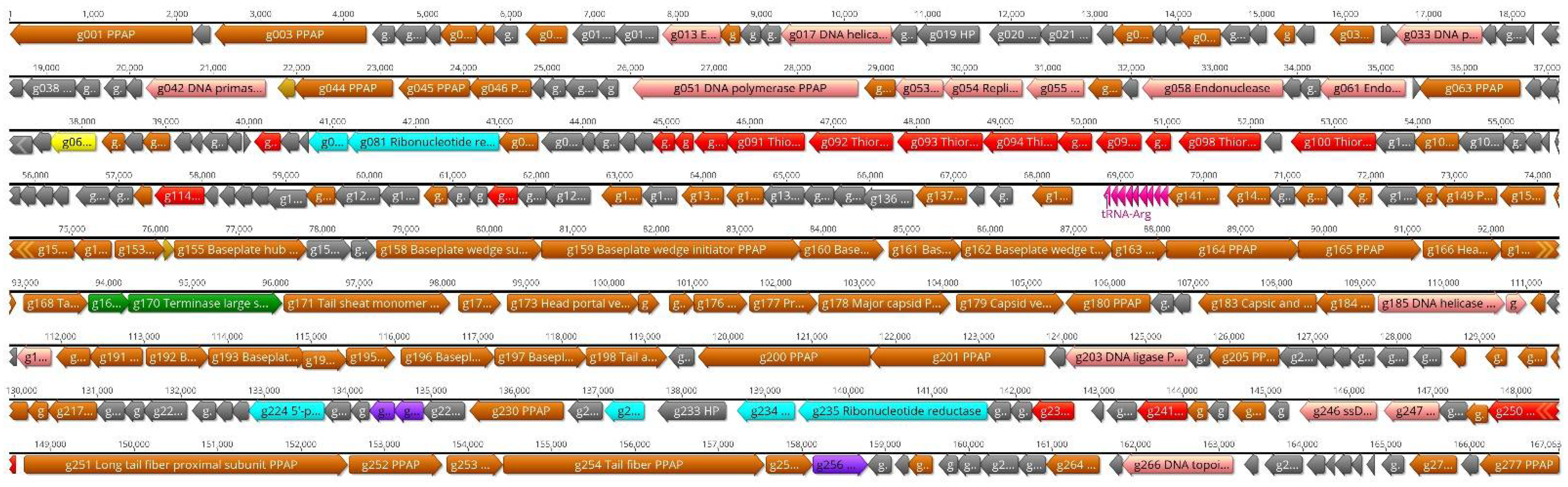
3.6. The Genome and Taxonomic Position of fD1
3.7. The Proteome of fD1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perry, R.D.; Fetherston, J.D. Yersinia pestis—Etiologic agent of plague. Clin. Microbiol. Rev. 1997, 10, 35–66. [Google Scholar] [CrossRef]
- Mead, P.S. Plague in Madagascar—A Tragic Opportunity for Improving Public Health. N. Engl. J. Med. 2018, 378, 106–108. [Google Scholar] [CrossRef]
- Nguyen, V.K.; Parra-Rojas, C.; Hernandez-Vargas, E.A. The 2017 plague outbreak in Madagascar: Data descriptions and epidemic modelling. Epidemics 2018, 25, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, B.J.; Rosso, M.L.; Schwan, T.G.; Carniel, E. High-frequency conjugative transfer of antibiotic resistance genes to Yersinia pestis in the flea midgut. Mol. Microbiol. 2002, 46, 349–354. [Google Scholar] [CrossRef]
- Filippov, A.A.; Sergueev, K.V.; He, Y.; Huang, X.-Z.; Gnade, B.T.; Mueller, A.J.; Fernandez-Prada, C.M.; Nikolich, M.P. Bacteriophage Therapy of Experimental Bubonic Plague in Mice. Adv. Exp. Med. Biol. 2012, 954, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Skurnik, M. Bacteriophages of Yersinia pestis. Adv. Exp. Med. Biol. 2016, 918, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.C. Cholera and plague in India: The bacteriophage inquiry of 1927–1936. J. Hist. Med. Allied Sci. 1993, 48, 275–301. [Google Scholar] [CrossRef] [Green Version]
- Garcia, E.; Elliott, J.M.; Ramanculov, E.; Chain, P.S.; Chu, M.C.; Molineux, I.J. The genome sequence of Yersinia pestis bacteriophage fA1122 reveals an intimate history with the coliphage T3 and T7 genomes. J. Bacteriol. 2003, 185, 5248–5262. [Google Scholar] [CrossRef] [Green Version]
- Filippov, A.A.; Sergueev, K.V.; He, Y.; Nikolich, M.P. Bacteriophages Capable of Lysing Yersinia pestis and Yersinia pseudotuberculosis: Efficiency of Plating Tests and Identification of Receptors in Escherichia coli K-12. Adv. Exp. Med. Biol. 2012, 954, 123–134. [Google Scholar] [CrossRef]
- Knapp, W. On the varying behavior of Pasteurella phages. Zent. Bakteriol. Orig. 1963, 190, 39–46. [Google Scholar]
- Filippov, A.A.; Sergueev, K.V.; He, Y.; Huang, X.-Z.; Gnade, B.T.; Mueller, A.J.; Fernandez-Prada, C.M.; Nikolich, M.P. Bacteriophage-Resistant Mutants in Yersinia pestis: Identification of Phage Receptors and Attenuation for Mice. PLoS ONE 2011, 6, e25486. [Google Scholar] [CrossRef] [Green Version]
- Schwudke, D.; Ergin, A.; Michael, K.; Volkmar, S.; Appel, B.; Knabner, D.; Konietzny, A.; Strauch, E. Broad-Host-Range Yersinia Phage PY100: Genome Sequence, Proteome Analysis of Virions, and DNA Packaging Strategy. J. Bacteriol. 2008, 190, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Xi, H.; Dai, J.; Zhong, Y.; Lu, S.; Wang, T.; Yang, L.; Guan, Y.; Wang, P. The characteristics and genome analysis of the novel Y. pestis phage JC221. Virus Res. 2020, 283, 197982. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [Green Version]
- Nolan, J.M.; Petrov, V.; Bertrand, C.; Krisch, H.M.; Karam, J.D. Genetic diversity among five T4-like bacteriophages. Virol. J. 2006, 3, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch. Virol. 2016, 161, 2921–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comeau, A.M.; Tremblay, D.; Moineau, S.; Rattei, T.; Kushkina, A.I.; Tovkach, F.I.; Krisch, H.M.; Ackermann, H.-W. Phage Morphology Recapitulates Phylogeny: The Comparative Genomics of a New Group of Myoviruses. PLoS ONE 2012, 7, e40102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuike, M.; Nishiki, I.; Iwasaki, Y.; Nakamura, Y.; Fujiwara, A.; Sugaya, E.; Kawato, Y.; Nagai, S.; Kobayashi, T.; Ototake, M.; et al. Full-genome sequence of a novel myovirus, GF-2, infecting Edwardsiella tarda: Comparison with other Edwardsiella myoviral genomes. Arch. Virol. 2015, 160, 2129–2133. [Google Scholar] [CrossRef]
- Hargreaves, K.R.; Clokie, M.R.J. A Taxonomic Review of Clostridium difficile Phages and Proposal of a Novel Genus, “Phimmp04likevirus”. Viruses 2015, 7, 2534–2541. [Google Scholar] [CrossRef] [Green Version]
- Garcia, E.; Nedialkov, Y.A.; Elliott, J.; Motin, V.; Brubaker, R.R. Molecular Characterization of KatY (Antigen 5), a Thermoregulated Chromosomally Encoded Catalase-Peroxidase of Yersinia pestis. J. Bacteriol. 1999, 181, 3114–3122. [Google Scholar] [CrossRef] [Green Version]
- Kiljunen, S.; Datta, N.; Dentovskaya, S.V.; Anisimov, A.P.; Knirel, Y.A.; Bengoechea, J.A.; Holst, O.; Skurnik, M. Identification of the lipopolysaccharide core of Yersinia pestis and Yersinia pseudotuberculosis as the receptor for bacteriophage fA1122. J. Bacteriol. 2011, 193, 4963–4972. [Google Scholar] [CrossRef] [Green Version]
- Portnoy, D.A.; Falkow, S. Virulence-associated plasmids from Yersinia enterocolitica and Yersinia pestis. J. Bacteriol. 1981, 148, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Tsubokura, M.; Aleksić, S. A simplified antigenic scheme for serotyping of Yersinia pseudotuberculosis: Phenotypic characterization of reference strains and preparation of O and H factor sera. Contrib. Microbiol. Immunol. 1995, 13, 99–105. [Google Scholar]
- Kenyon, J.J.; Duda, K.A.; De Felice, A.; Cunneen, M.M.; Molinaro, A.; Laitinen, J.; Skurnik, M.; Holst, O.; Reeves, P.; De Castro, C. Serotype O:8 isolates in the Yersinia pseudotuberculosis complex have different O-antigen gene clusters and produce various forms of rough LPS. Innate Immun. 2016, 22, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Skurnik, M. Studies on the Virulence Plasmids of Yersinia Species. Ph.D. Thesis, University of Oulu, Oulu, Finland, 1985. [Google Scholar]
- Macrina, F.L.; Kopecko, D.J.; Jones, K.R.; Ayers, D.J.; McCowen, S.M. A multiple plasmid-containing Escherichia coli strain: Convenient source of size reference plasmid molecules. Plasmid 1978, 1, 417–420. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Sambrook, J.; Russell, D.W. Purification of bacteriophage λ particles by centrifugation through a glycerol step gradient. CSH Protoc. 2006. [Google Scholar] [CrossRef]
- Staden, R.; Judge, D.P.; Bonfield, J.K. Managing sequencing projects in the GAP4 environment. In Introduction to Bioinformatics. A Theoretical and Practical Approach; Krawetz, S.A., Womble, D.D., Eds.; Humana Press: Totowa, NJ, USA, 2003; pp. 327–344. [Google Scholar]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.-A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.J.; Berriman, M.; Tivey, A.; Patel, C.; Böhme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.-A. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carver, T.; Harris, S.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2011, 28, 464–469. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Kallio, M.A.; Tuimala, J.T.; Hupponen, T.; Klemelä, P.; Gentile, M.; Scheinin, I.; Koski, M.; Käki, J.; Korpelainen, E.I. Chipster: User-friendly analysis software for microarray and other high-throughput data. BMC Genom. 2011, 12, 507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Asaf, S. Automatic annotation of microbial genomes and metagenomic sequences. Metagenomics and its applications. In Agriculture, Biomedicine and Environmental Studies; Li, R.W., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2010; pp. 61–78. [Google Scholar]
- Teleman, J.; Dowsey, A.; Gonzalez-Galarza, F.F.; Perkins, S.; Pratt, B.; Röst, H.; Malmström, L.; Malmström, J.; Jones, A.; Deutsch, E.W.; et al. Numerical Compression Schemes for Proteomics Mass Spectrometry Data. Mol. Cell. Proteom. 2014, 13, 1537–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Bauch, A.; Adamczyk, I.; Buczek, P.; Elmer, F.-J.; Enimanev, K.; Glyzewski, P.; Kohler, M.; Pylak, T.; Quandt, A.; Ramakrishnan, C.; et al. openBIS: A flexible framework for managing and analyzing complex data in biology research. BMC Bioinform. 2011, 12, 468. [Google Scholar] [CrossRef] [Green Version]
- Craig, R.; Beavis, R.C. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 2003, 17, 2310–2316. [Google Scholar] [CrossRef]
- Keller, A.; Nesvizhskii, A.; Kolker, E.; Aebersold, R. Empirical Statistical Model to Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Ackermann, H.W.; Krisch, H.M.; Comeau, A.M. Morphology and genome sequence of phage φ1402: A dwarf myovirus of the predatory bacterium Bdellovibrio bacteriovorus. Bacteriophage 2011, 1, 138–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuike, M.; Sugaya, E.; Nakamura, Y.; Shigenobu, Y.; Kawato, Y.; Kai, W.; Nagai, S.; Fujiwara, A.; Sano, M.; Kobayashi, T.; et al. Complete Genome Sequence of a Novel Myovirus which Infects Atypical Strains of Edwardsiella tarda. Genome Announc. 2013, 1, e00248-12. [Google Scholar] [CrossRef] [Green Version]
- Fokine, A.; Chipman, P.R.; Leiman, P.; Mesyanzhinov, V.V.; Rao, V.; Rossmann, M.G. Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. USA 2004, 101, 6003–6008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostyuchenko, V.A.; Chipman, P.R.; Leiman, P.G.; Arisaka, F.; Mesyanzhinov, V.V.; Rossmann, M.G. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat. Struct. Mol. Biol. 2005, 12, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, C.; Caumont-Sarcos, A.; Comeau, A.M.; Krisch, H.M. Isolation and genomic characterization of the first phage infecting Iodobacteria: ϕPLPE, a myovirus having a novel set of features. Environ. Microbiol. Rep. 2009, 1, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.M.; Brown, J.M.; Sharma, R.S.; VanInsberghe, D.; Elsherbini, J.; Polz, M.; Kelly, L. Viruses of the Nahant Collection, characterization of 251 marine Vibrionaceae viruses. Sci. Data 2018, 5, 180114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, T.; Couillault, C.; Rockenfeller, P.; Boucrot, E.; Dumont, A.; Schroeder, N.; Hermant, A.; Knodler, L.; Lécine, P.; Steele-Mortimer, O.; et al. The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc. Natl. Acad. Sci. USA 2006, 103, 13497–13502. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.; Pajunen, M.; Jun, J.; Skurnik, M. T4-like Bacteriophages Isolated from Pig Stools Infect Yersinia pseudotuberculosis and Yersinia pestis Using LPS and OmpF as Receptors. Viruses 2021, 13, 296. [Google Scholar] [CrossRef]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc. Natl. Acad. Sci. USA 2015, 112, E4919–E4928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunne, M.; Denyes, J.M.; Arndt, H.; Loessner, M.J.; Leiman, P.; Klumpp, J. Salmonella Phage S16 Tail Fiber Adhesin Features a Rare Polyglycine Rich Domain for Host Recognition. Structure 2018, 26, 1573–1582.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimov, A.P.; Dentovskaya, S.V.; Kondakova, A.N.; Lindner, B.; Shaikhutdinova, R.Z.; Kocharova, N.A.; Senchenkova, S.N.; Knirel, Y.A. Yersinia pestis lipopolysaccharide in host-pathogen interactions. In The Challenge of Highly Pathogenic Microorganisms; Springer: Berlin/Heidelberg, Germany, 2010; pp. 77–87. [Google Scholar]
- Bengoechea, J.A.; Najdenski, H.; Skurnik, M. Lipopolysaccharide O antigen status of Yersinia enterocolitica O:8 is essential for virulence and absence of O antigen affects the expression of other Yersinia virulence factors. Mol. Microbiol. 2004, 52, 451–469. [Google Scholar] [CrossRef] [PubMed]
- Najdenski, H.; Golkocheva, E.; Vesselinova, A.; Bengoechea, J.; Skurnik, M. Proper expression of the O-antigen of lipopolysaccharide is essential for the virulence of Yersinia enterocolitica O:8 in experimental oral infection of rabbits. FEMS Immunol. Med. Microbiol. 2003, 38, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Skurnik, M. Molecular genetics, biochemistry and biological role of Yersinia lipopolysaccharide. In The Genus Yersinia: Entering the Functional Genomic Era; Skurnik, M., Granfors, K., Bengoechea, J.A., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2003; pp. 187–189. [Google Scholar]
- Skurnik, M.; Venho, R.; Bengoechea, J.; Moriyon, I. The lipopolysaccharide outer core of Yersinia enterocolitica serotype O:3 is required for virulence and plays a role in outer membrane integrity. Mol. Microbiol. 1999, 31, 1443–1462. [Google Scholar] [CrossRef]
- Darwin, A.; Miller, V.L. Identification of Yersinia enterocolitica genes affecting survival in an animal host using signature-tagged transposon mutagenesis. Mol. Microbiol. 1999, 32, 51–62. [Google Scholar] [CrossRef]
- Karlyshev, A.V.; Oyston, P.C.F.; Williams, K.; Clark, G.C.; Titball, R.W.; Winzeler, E.A.; Wren, B.W. Application of High-Density Array-Based Signature-Tagged Mutagenesis to Discover Novel Yersinia Virulence-Associated Genes. Infect. Immun. 2001, 69, 7810–7819. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strains | Skurnik Lab STORAGE # | EOP of fEV-1 | EOP of fD1 | Comments | Reference |
|---|---|---|---|---|---|
| Yersinia pestis | |||||
| KIM D27 | 1418 | 1 | 1 | Non-pigmented derivative of wild type strain KIM10. Lcr+ Pgm− Pst+ | [20] |
| KIM D27-ΔwaaQ | 5147 | 10−4 | 1 | ΔwaaQ::nptII KanR, deep rough LPS missing distal N-acetylglucosamine and two distal heptose residues | [21] |
| KIM D27-ΔwaaE | 5149 | 10−5 | 1 | ΔwaaE::nptII KanR, deep rough LPS missing two distal heptose residues and the proximal glucose residue | [21] |
| KIM D27-ΔwaaL | 5150 | 1 | 1 | ΔwaaL::nptII KanR, deep rough LPS missing the distal N-acetylglucosamine residue | [21] |
| KIM D27-ΔwabD | 5151 | 1 | 1 | ΔwabD::nptII KanR, rough variant LPS missing distal Gal residue | [21] |
| EV76 | 1281 | 1 | 1 | Non-pigmented derivative of wild type strain EV | [22] |
| Yersinia pseudotuberculosis | |||||
| Pa 3606 | 2061 | 0 | 0 | Serotype O:1b | [23] |
| 204 | 2069 | 1 | Serotype O:5a | [23] | |
| 197 | 2070 | 0 | 0 | Serotype O:5b | [23] |
| 151 | 2073 | 0 | 10−4 | Spontaneous rough derivative of serotype O:4a | [23,24] |
| YPIII::Δwb | 2648 | 0 | 0 | [21] | |
| PB1::Δwb | 2649 | 0 | 10−3 | [21] | |
| Escherichia coli | |||||
| ME 2881-2 | ld 536 | 0 | 0 | Clinical human isolate | |
| ME 3128 | ld 537 | 0 | 0 | Clinical human isolate | |
| ME 2886-2 | 6588 | 0 | 1 | Clinical human isolate | |
| TS 2239-1 | 6729 | 0 | 0 | Clinical human isolate | |
| ME 2861 | ld 541 | 0 | 0 | Clinical human isolate | |
| ME 2863 | ld 542 | 0 | 0 | Clinical human isolate | |
| TS 2174 | ld 543 | 0 | 0 | Clinical human isolate | |
| TS 2757 | ld 548 | 0 | 0 | Clinical human isolate | |
| ME 2671-1 | ld 558 | 0 | 0 | Clinical human isolate | |
| ME 2676-1 | ld 559 | 0 | 0 | Clinical human isolate | |
| ME 2680-1 | 6589 | 0 | 1 | Clinical human isolate | |
| ME 2683-1 | 6590 | 0 | 1 | Clinical human isolate | |
| US 1439 | 6500 | 0 | 0 | Clinical human isolate | |
| KP 1708 | 6501 | 0 | 0 | Clinical human isolate | |
| ME 1658 | 6503 | 0 | 0 | Clinical human isolate | |
| ME 1920 | 6504 | 0 | 0 | Clinical human isolate | |
| US 1769-2 | 6508 | 0 | 0 | Clinical human isolate | |
| 1100 (R1 drd-19k-1) | 251 | 0 | 1 | Laboratory strain | [25] |
| C600 su (lambda cI857) | 253 | 0 | 1 | Laboratory strain | |
| V517 | 258 | 0 | 1 | Clinical isolate | [26] |
| RY13 | 449 | 0 | 1 | Laboratory strain | |
| LE392 (P1-cml, clr100) | 629 | 0 | 1 | Laboratory strain | |
| P678-54 | 630 | 0 | 1 | Laboratory strain | |
| LE392 | 631 | 0 | 1 | Laboratory strain | |
| JM103 | 1247 | 0 | 1 | Laboratory strain | |
| PM191 | 1266 | 0 | 1 | Laboratory strain | |
| D21f2 | 1354 | 0 | 0.1 | Laboratory strain | |
| D21 | 1355 | 0 | 1 | Laboratory strain | |
| DH1 | 1378 | 0 | 1 | Laboratory strain | |
| HB101 | 1389 | 0 | 1 | Laboratory strain | |
| C600 | 1424 | 0 | 1 | Laboratory strain | |
| Klebsiella oxytoca | |||||
| TS 2752 | 547 | 0 | 0 | Clinical human isolate | |
| Shigella | |||||
| 872 | 707 | 0 | 0 | Quality control strain | |
| Primer | Sequence (5′-3′) | Position in LT992259 |
|---|---|---|
| fEV1-R | CCTTGCTTGCATTCAGTTCA | 1198..1179 |
| fEV1-R2 | TGCACCTTCATTTCAAGCAG | 580..561 |
| fEV1-R3 | GCTGAAGTATCGGCTTCCAG | 449..430 |
| fEV1-F | GAAGGAGATAGTGCGCGTTC | 37351..37370 |
| fEV1-F2 | GTGAAACGCTTGATGCTGAA | 38002..38021 |
| fEV1-F3 | ACCGCACATTCAACAAAACA | 38114..38133 |
| fEV1-F4 | TCGCCTTCAGGGTATCAATC | 33590..33609 |
| fEV1-R4 | TCAAGACCCATTGCACTGAA | 34155..34136 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skurnik, M.; Jaakkola, S.; Mattinen, L.; von Ossowski, L.; Nawaz, A.; Pajunen, M.I.; Happonen, L.J. Bacteriophages fEV-1 and fD1 Infect Yersinia pestis. Viruses 2021, 13, 1384. https://doi.org/10.3390/v13071384
Skurnik M, Jaakkola S, Mattinen L, von Ossowski L, Nawaz A, Pajunen MI, Happonen LJ. Bacteriophages fEV-1 and fD1 Infect Yersinia pestis. Viruses. 2021; 13(7):1384. https://doi.org/10.3390/v13071384
Chicago/Turabian StyleSkurnik, Mikael, Salla Jaakkola, Laura Mattinen, Lotta von Ossowski, Ayesha Nawaz, Maria I. Pajunen, and Lotta J. Happonen. 2021. "Bacteriophages fEV-1 and fD1 Infect Yersinia pestis" Viruses 13, no. 7: 1384. https://doi.org/10.3390/v13071384
APA StyleSkurnik, M., Jaakkola, S., Mattinen, L., von Ossowski, L., Nawaz, A., Pajunen, M. I., & Happonen, L. J. (2021). Bacteriophages fEV-1 and fD1 Infect Yersinia pestis. Viruses, 13(7), 1384. https://doi.org/10.3390/v13071384