
CARs—A New Perspective to HCMV Treatment


Abstract
:1. Evasion of the Host Immune System by HCMV
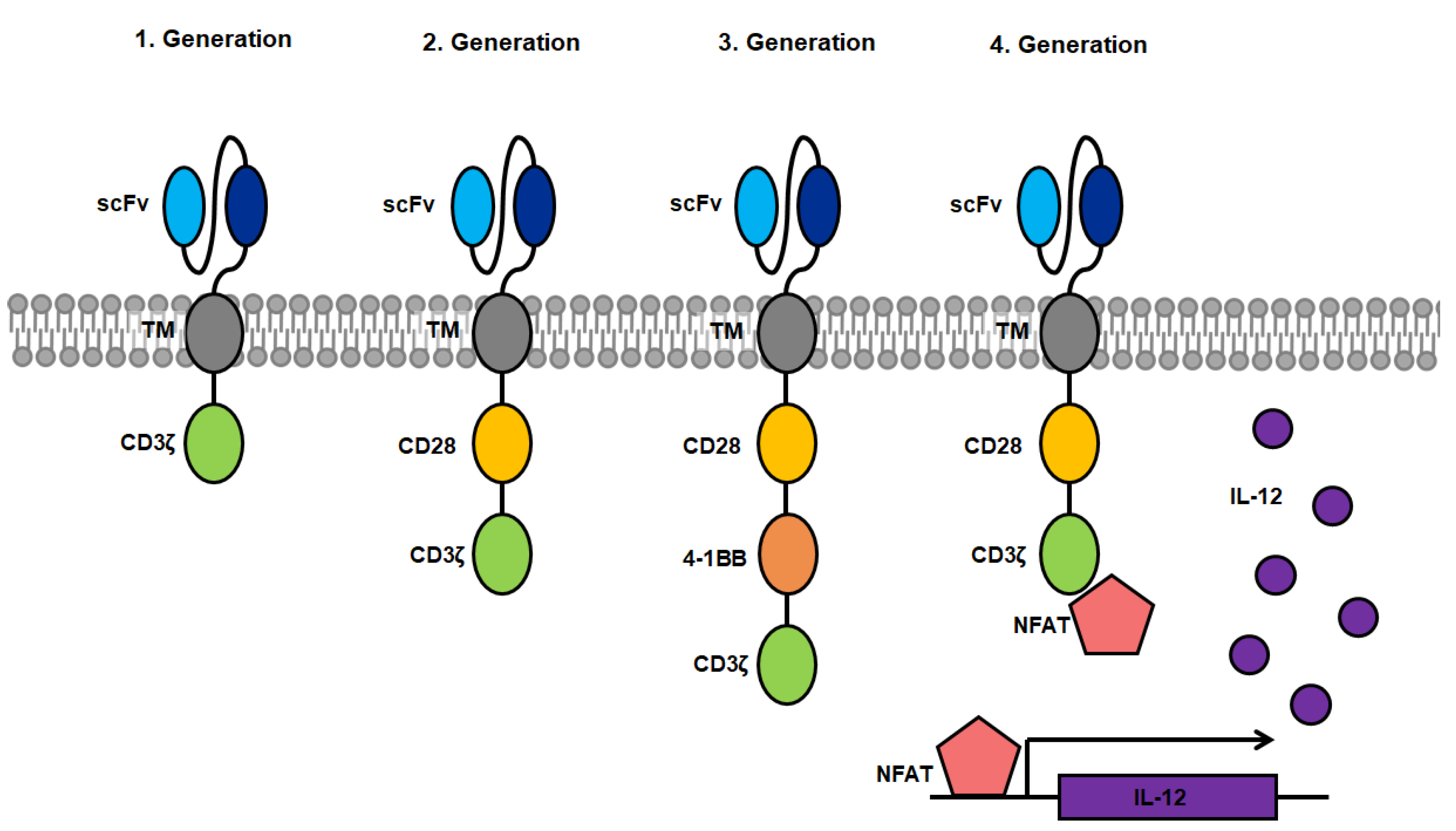
2. History of CARs
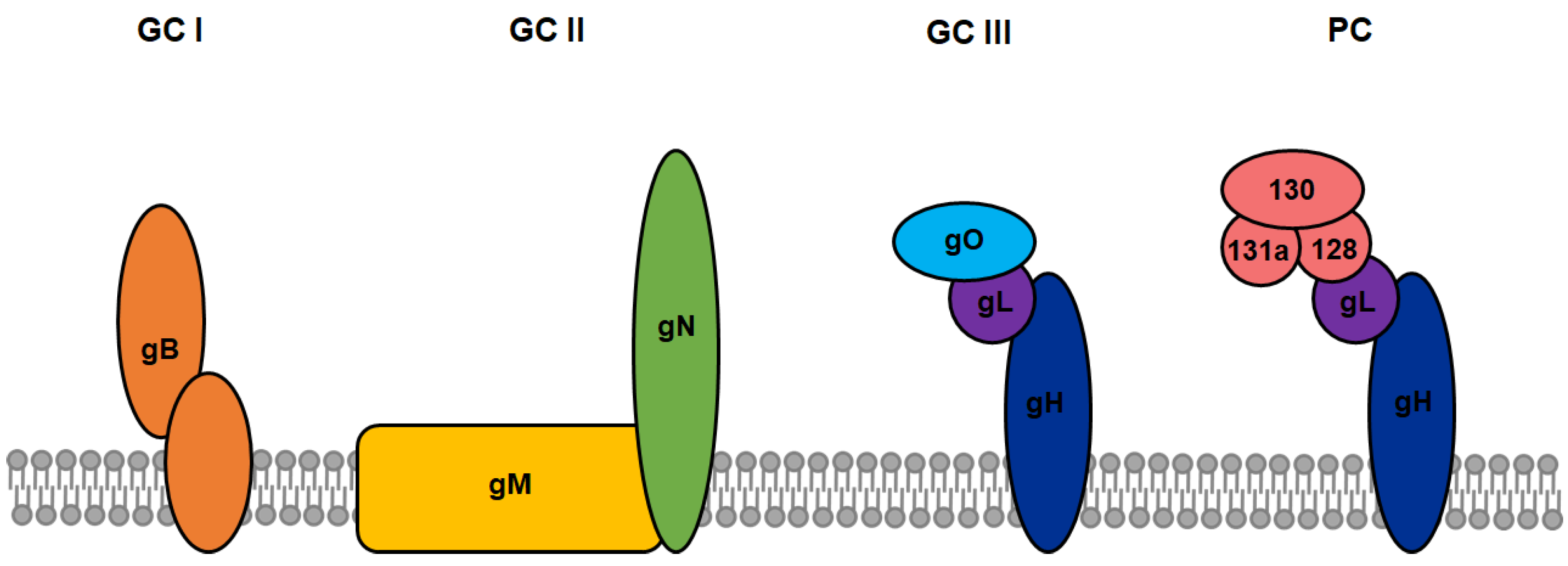
3. Viral Glycoproteins as Markers of Infected Cells
4. HCMV-Specific CARs
5. Discussion of Other Antiviral CARs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boeckh, M.; Ljungman, P. How we treat cytomegalovirus in hematopoietic cell transplant recipients. Blood 2009, 113, 5711–5719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert Koch Institut. Zytomegalievirus-Infektion: RKI Ratgeber. Available online: https://www.rki.de/DE/Content/Infekt/EpidBull/Merkblaetter/Ratgeber_Zytomegalievirus.html (accessed on 30 June 2021).
- Majeed, A.; Latif, A.; Kapoor, V.; Sohail, A.; Florita, C.; Georgescu, A.; Zangeneh, T. Resistant Cytomegalovirus Infection in Solid-organ Transplantation: Single-center Experience, Literature Review of Risk Factors, and Proposed Preventive Strategies. Transplant. Proc. 2018, 50, 3756–3762. [Google Scholar] [CrossRef]
- Helou, G.E.; Razonable, R.R. Letermovir for the prevention of cytomegalovirus infection and disease in transplant recipients: An evidence-based review. Infect. Drug Resist. 2019, 12, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Crough, T.; Khanna, R. Immunobiology of human cytomegalovirus: From bench to bedside. Clin. Microbiol. Rev. 2009, 22, 76–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halenius, A.; Gerke, C.; Hengel, H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: So many targets—but how many arrows in the quiver? Cell. Mol. Immunol. 2015, 12, 139–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrales-Aguilar, E.; Hoffmann, K.; Hengel, H. CMV-encoded Fcγ receptors: Modulators at the interface of innate and adaptive immunity. Semin. Immunopathol. 2014, 36, 627–640. [Google Scholar] [CrossRef]
- Riddell, S.R.; Watanabe, K.S.; Goodrich, J.M.; Li, C.R.; Agha, M.E.; Greenberg, P.D. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992, 257, 238–241. [Google Scholar] [CrossRef]
- Riddell, S.R.; Walter, B.A.; Gilbert, M.J.; Greenberg, P.D. Selective reconstitution of CD8+ cytotoxic T lymphocyte responses in immunodeficient bone marrow transplant recipients by the adoptive transfer of T cell clones. Bone Marrow Transplant. 1994, 14, S78–S84. [Google Scholar] [PubMed]
- Tzannou, I.; Watanabe, A.; Naik, S.; Daum, R.; Kuvalekar, M.; Leung, K.S.; Martinez, C.; Sasa, G.; Wu, M.; Gee, A.P.; et al. “Mini” bank of only 8 donors supplies CMV-directed T cells to diverse recipients. Blood Adv. 2019, 3, 2571–2580. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [PubMed]
- Brocker, T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood 2000, 96, 1999–2001. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.F.; Latouche, J.B.; Tan, C.; Cheung, N.K.; Sadelain, M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [Green Version]
- van der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Feldman, S.A.; Zhao, Y.; Xu, H.; Black, M.A.; Morgan, R.A.; Wilson, W.H.; Rosenberg, S.A. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 2009, 32, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Food & Drug Administration, KYMRIAH (Tisagenlecleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel (accessed on 30 June 2021).
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor—Modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [Green Version]
- Enblad, G.; Karlsson, H.; Gammelgård, G.; Wenthe, J.; Lövgren, T.; Amini, R.M.; Wikstrom, K.I.; Essand, M.; Savoldo, B.; Hallböök, H.; et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res. 2018, 24, 6185–6194. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef]
- Landgraf, K.E.; Williams, S.R.; Steiger, D.; Gebhart, D.; Lok, S.; Martin, D.W.; Roybal, K.T.; Kim, K.C. convertibleCARs: A chimeric antigen receptor system for flexible control of activity and antigen targeting. Commun. Biol. 2020, 3, 296. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar] [CrossRef]
- Bootz, A.; Karbach, A.; Spindler, J.; Kropff, B.; Reuter, N.; Sticht, H.; Winkler, T.H.; Britt, W.J.; Mach, M. Protective capacity of neutralizing and non-neutralizing antibodies against glycoprotein B of cytomegalovirus. PLoS Pathog. 2017, 13, e1006601. [Google Scholar] [CrossRef]
- Karbach, A. Protection from Cytomegalovirus Infection by Glycoprotein-specific Monoclonal Antibodies: Schutz vor Infektion mit dem Cytomegalievirus durch glykoproteinspezifische monoklonale Antikörper. Ph.D. Thesis, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany, 2012. [Google Scholar]
- Smuda, C.; Bogner, E.; Radsak, K. The human cytomegalovirus glycoprotein B gene (ORF UL55) is expressed early in the infectious cycle. J. Gen. Virol. 1997, 78, 1981–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodrum, F. Human Cytomegalovirus Latency: Approaching the Gordian Knot. Annu. Rev. Virol. 2016, 3, 333–357. [Google Scholar] [CrossRef] [Green Version]
- Full, F.; Lehner, M.; Thonn, V.; Goetz, G.; Scholz, B.; Kaufmann, K.B.; Mach, M.; Abken, H.; Holter, W.; Ensser, A. T cells engineered with a cytomegalovirus-specific chimeric immunoreceptor. J. Virol. 2010, 84, 4083–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proff, J.; Walterskirchen, C.; Brey, C.; Geyeregger, R.; Full, F.; Ensser, A.; Lehner, M.; Holter, W. Cytomegalovirus-Infected Cells Resist T Cell Mediated Killing in an HLA-Recognition Independent Manner. Front. Microbiol. 2016, 7, 844. [Google Scholar] [CrossRef] [PubMed]
- Delmas, S.; Brousset, P.; Clément, D.; Le Roy, E.; Davignon, J.-L. Anti-IE1 CD4+ T-cell clones kill peptide-pulsed, but not human cytomegalovirus-infected, target cells. J. Gen. Virol. 2007, 88, 2441–2449. [Google Scholar] [CrossRef]
- Halle, S.; Keyser, K.A.; Stahl, F.R.; Busche, A.; Marquardt, A.; Zheng, X.; Galla, M.; Heissmeyer, V.; Heller, K.; Boelter, J.; et al. In Vivo Killing Capacity of Cytotoxic T Cells Is Limited and Involves Dynamic Interactions and T Cell Cooperativity. Immunity 2016, 44, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J.; Weiland, F.; Münch, K.; Jonjic, S.; Lüske, A.; Koszinowski, U.H. Interstitial murine cytomegalovirus pneumonia after irradiation: Characterization of cells that limit viral replication during established infection of the lungs. J. Virol. 1985, 55, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J.; Mutter, W.; Münch, K.; Bühring, H.J.; Koszinowski, U.H. CD8-positive T lymphocytes specific for murine cytomegalovirus immediate-early antigens mediate protective immunity. J. Virol. 1987, 61, 3102–3108. [Google Scholar] [CrossRef] [Green Version]
- Proff, J.; Brey, C.U.; Ensser, A.; Holter, W.; Lehner, M. Turning the tables on cytomegalovirus: Targeting viral Fc receptors by CARs containing mutated CH2-CH3 IgG spacer domains. J. Transl. Med. 2018, 16, 26. [Google Scholar] [CrossRef]
- Almåsbak, H.; Walseng, E.; Kristian, A.; Myhre, M.R.; Suso, E.M.; Munthe, L.A.; Andersen, J.T.; Wang, M.Y.; Kvalheim, G.; Gaudernack, G.; et al. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 2015, 22, 391–403. [Google Scholar] [CrossRef]
- Olbrich, H.; Theobald, S.J.; Slabik, C.; Gerasch, L.; Schneider, A.; Mach, M.; Shum, T.; Mamonkin, M.; Stripecke, R. Adult and Cord Blood-Derived High-Affinity gB-CAR-T Cells Effectively React Against Human Cytomegalovirus Infections. Hum. Gene Ther. 2020, 31, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Chiuppesi, F.; Nguyen, M.; Hausner, M.A.; Nguyen, J.; Kha, M.; Iniguez, A.; Wussow, F.; Diamond, D.J.; Yang, O.O. Chimeric Antigen Receptors Targeting Human Cytomegalovirus. J. Infect. Dis. 2020, 222, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zou, F.; Lu, L.; Chen, C.; He, D.; Zhang, X.; Tang, X.; Liu, C.; Li, L.; Zhang, H. Chimeric Antigen Receptor T Cells Guided by the Single-Chain Fv of a Broadly Neutralizing Antibody Specifically and Effectively Eradicate Virus Reactivated from Latency in CD4+ T Lymphocytes Isolated from HIV-1-Infected Individuals Receiving Suppressive Combined Antiretroviral Therapy. J. Virol. 2016, 90, 9712–9724. [Google Scholar] [CrossRef] [Green Version]
- Slabik, C.; Kalbarczyk, M.; Danisch, S.; Zeidler, R.; Klawonn, F.; Volk, V.; Krönke, N.; Feuerhake, F.; Ferreira de Figueiredo, C.; Blasczyk, R.; et al. CAR-T Cells Targeting Epstein-Barr Virus gp350 Validated in a Humanized Mouse Model of EBV Infection and Lymphoproliferative Disease. Mol. Ther. Oncolytics 2020, 18, 504–524. [Google Scholar] [CrossRef]
- Sautto, G.A.; Wisskirchen, K.; Clementi, N.; Castelli, M.; Diotti, R.A.; Graf, J.; Clementi, M.; Burioni, R.; Protzer, U.; Mancini, N. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut 2016, 65, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Bohne, F.; Chmielewski, M.; Ebert, G.; Wiegmann, K.; Kürschner, T.; Schulze, A.; Urban, S.; Krönke, M.; Abken, H.; Protzer, U. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology 2008, 134, 239–247. [Google Scholar] [CrossRef]
- Krebs, K.; Böttinger, N.; Huang, L.-R.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| CAR T-Cell Therapy | Antigen Specific T-Cell Therapy | |
|---|---|---|
| Antigen-specific target cell recognition | yes | yes |
| HLA-independent mode of action | yes | no |
| HLA-matching required | no | partial match |
| Seronegative donor possible | yes | no |
| Circumvention of CMV-mediated HLA-targeted immune evasion | yes | no |
| Isolation, ex vivo expansion and genetic modification of T-cells (mRNA or Lentiviral vector) | yes | no |
| Isolation and ex vivo expansion of specific T-cells required | no | yes |
| ‘Off-the-shelf’ therapy possible | yes | HLA dependent |
| Eradication of virus from host | no | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bednar, C.; Ensser, A. CARs—A New Perspective to HCMV Treatment. Viruses 2021, 13, 1563. https://doi.org/10.3390/v13081563
Bednar C, Ensser A. CARs—A New Perspective to HCMV Treatment. Viruses. 2021; 13(8):1563. https://doi.org/10.3390/v13081563
Chicago/Turabian StyleBednar, Christopher, and Armin Ensser. 2021. "CARs—A New Perspective to HCMV Treatment" Viruses 13, no. 8: 1563. https://doi.org/10.3390/v13081563
APA StyleBednar, C., & Ensser, A. (2021). CARs—A New Perspective to HCMV Treatment. Viruses, 13(8), 1563. https://doi.org/10.3390/v13081563