
The Endogenous RIG-I Ligand Is Generated in Influenza A-Virus Infected Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Kits and Reagents
2.2. Cells and Viruses
2.3. Light Microscopy
2.4. Luciferase Assay
2.5. PBMC Stimulation
2.6. RNA-Isolation and qRT-PCR
2.7. RNase A Digestion
2.8. Viral Infections
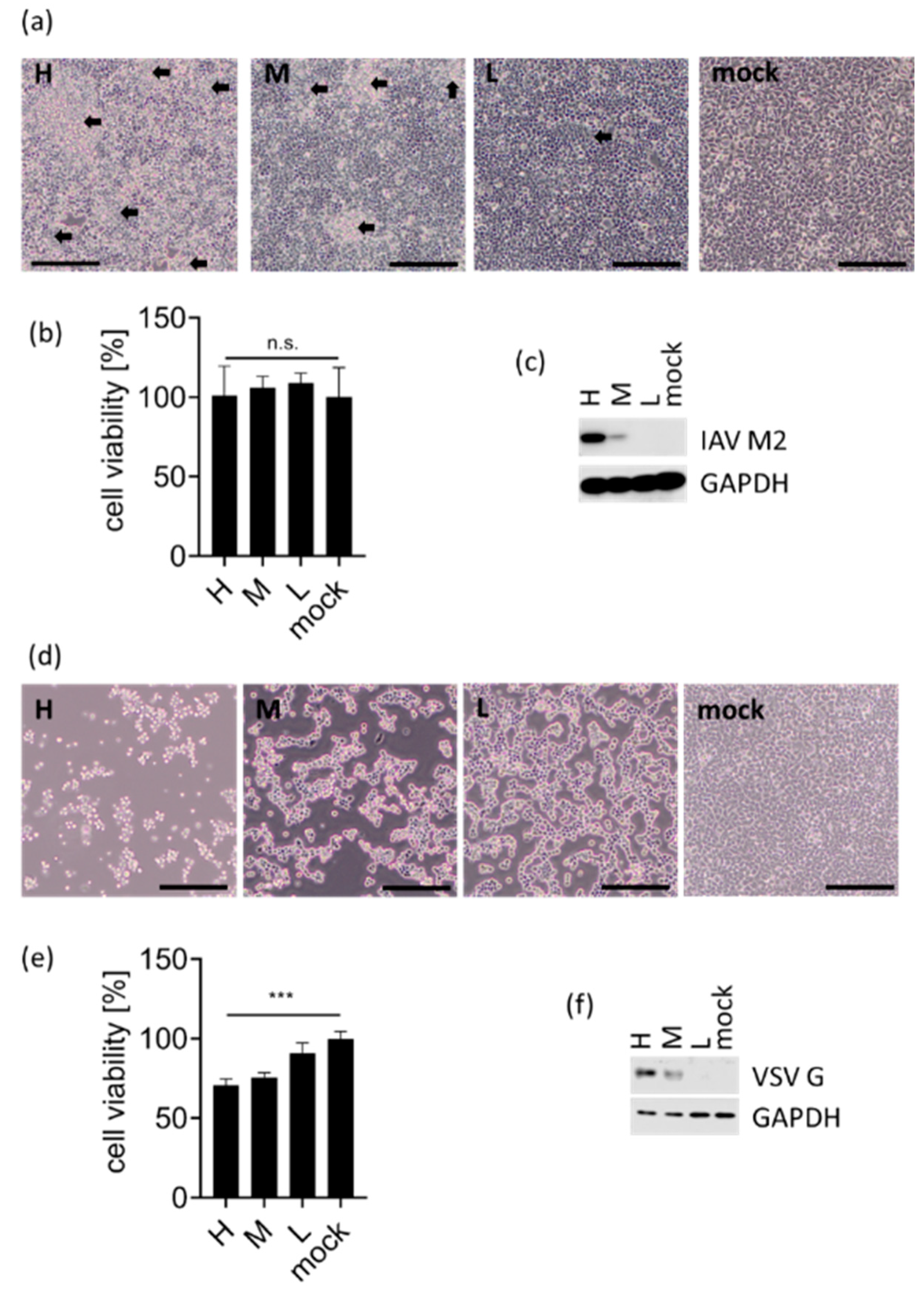
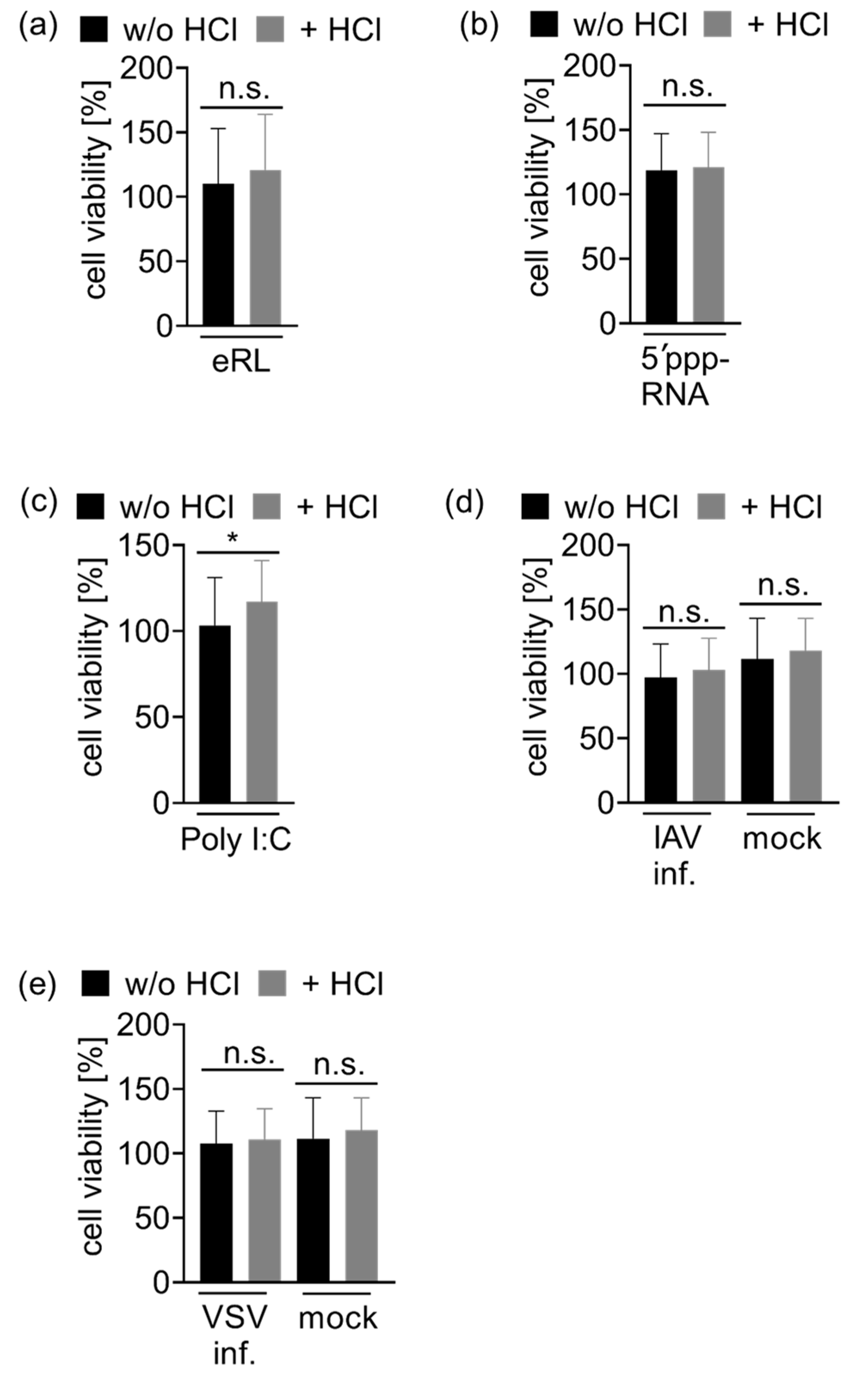
2.9. Cell Viability Assay
2.10. Western Blotting
2.11. Statistical Analysis
3. Results
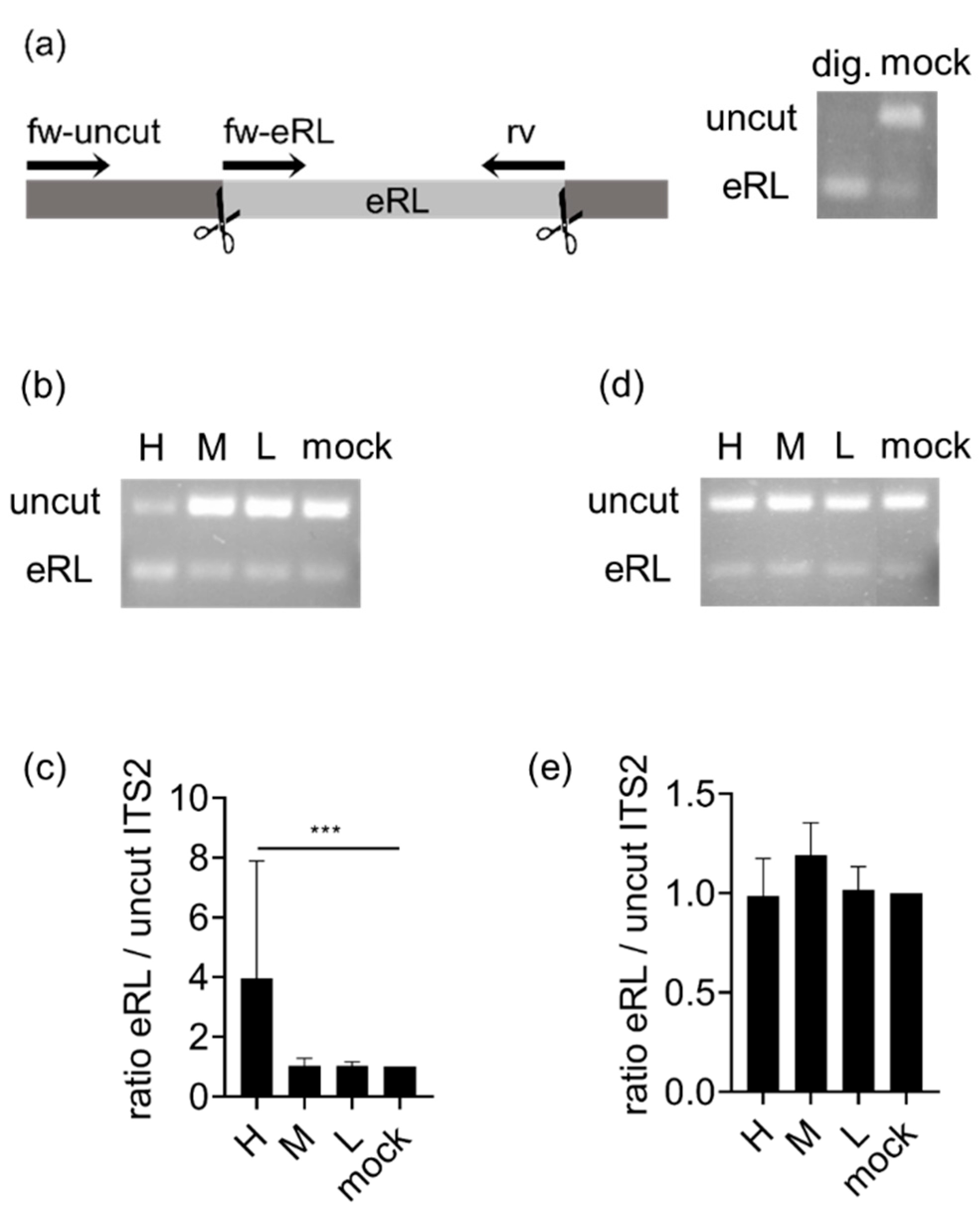
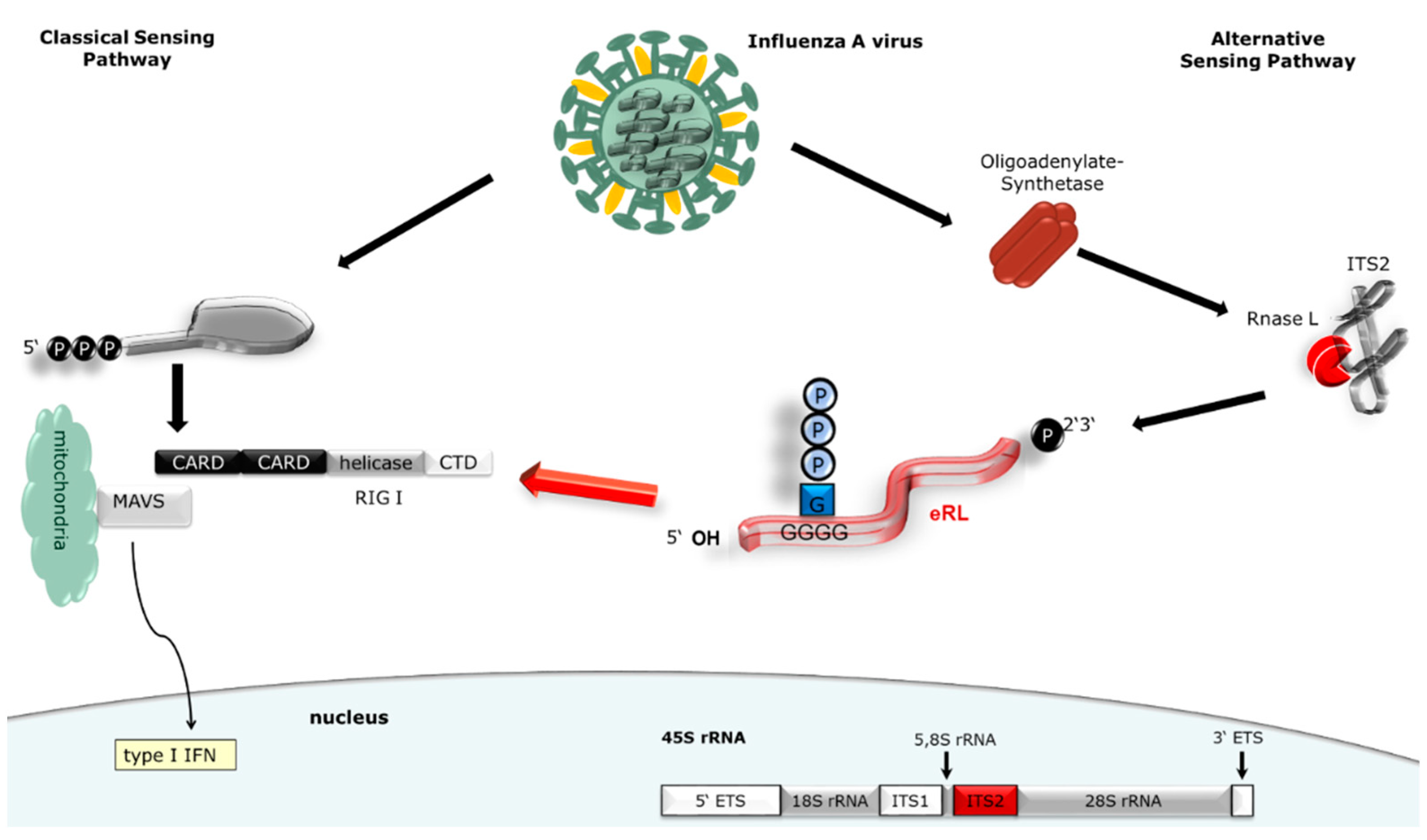
3.1. eRL Is Generated in IAV-Infected Cells
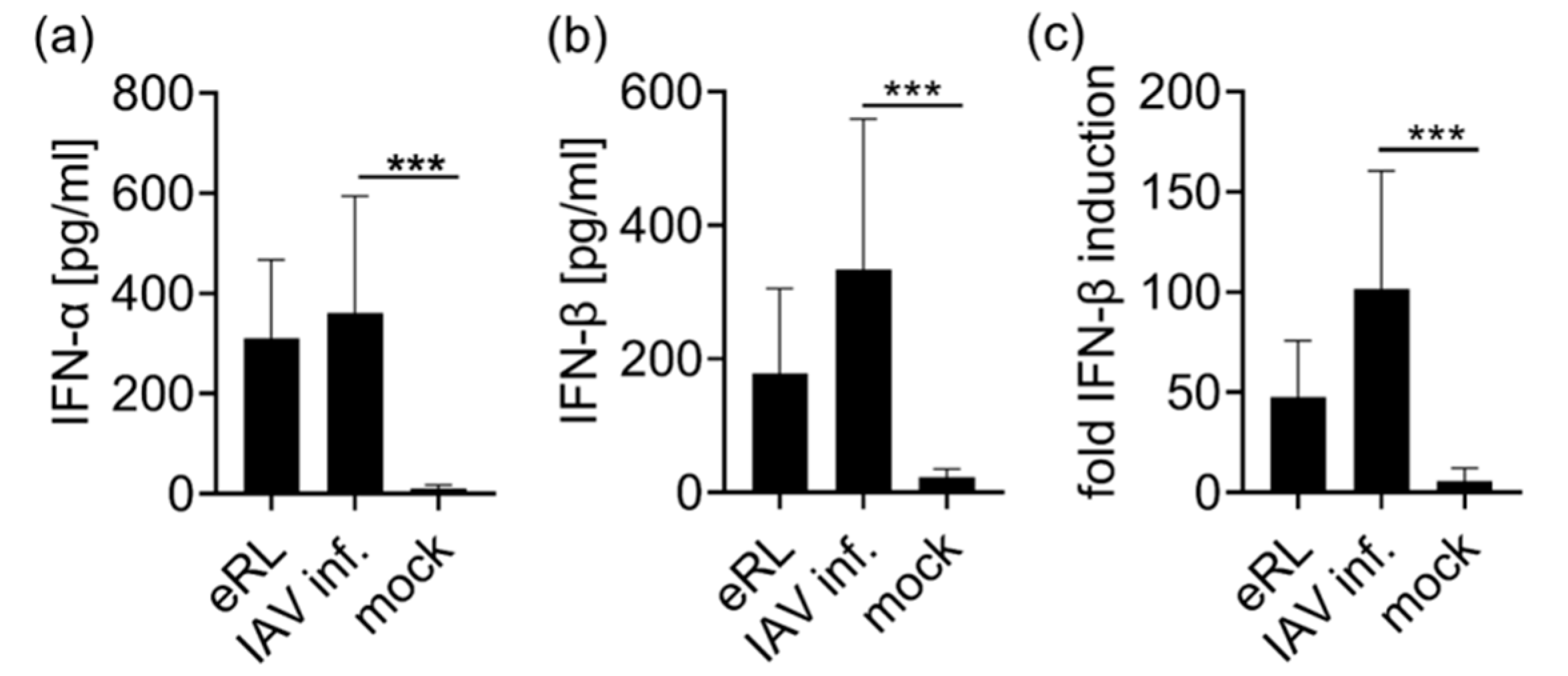
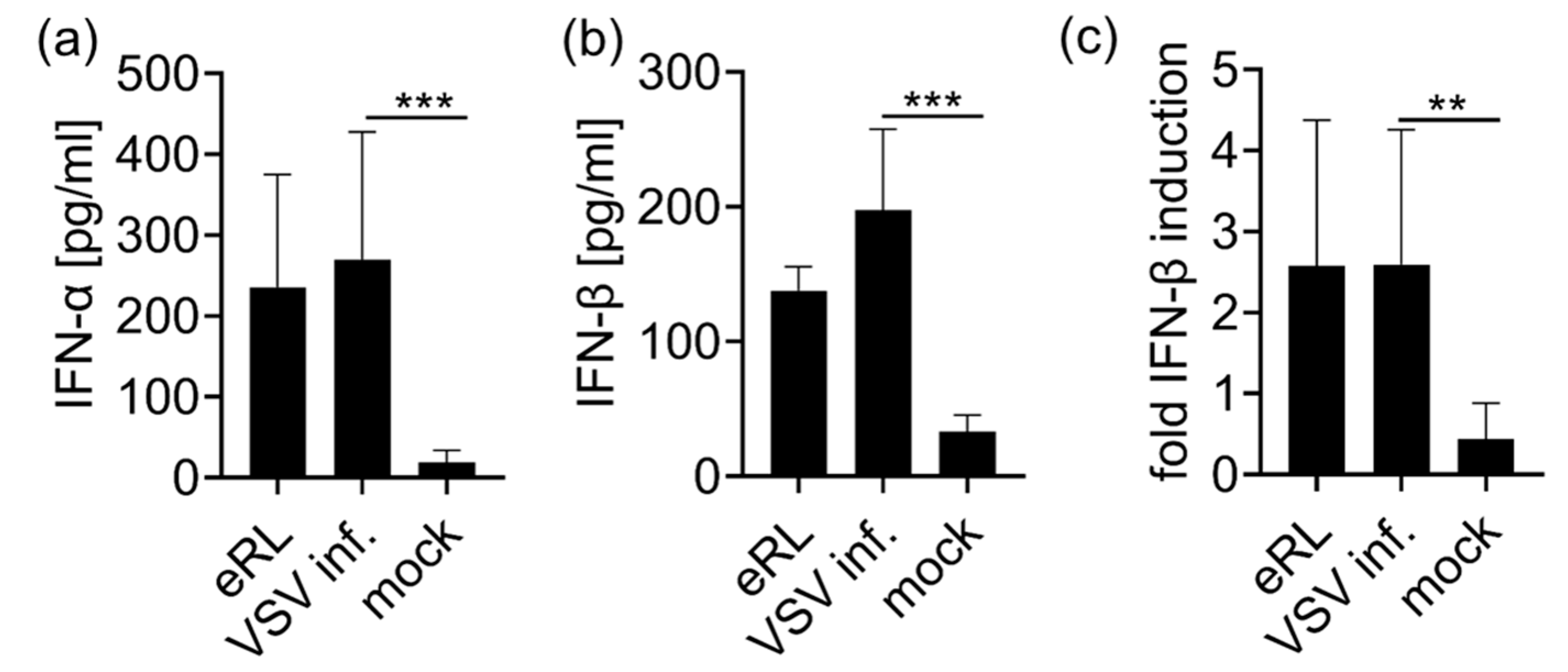
3.2. Virus-Induced RNA Species Stimulate Type I IFN Via RIG-I
3.3. IAV-Induced RNA Species Activate Type I IFN Release Independently of IAV Genomes
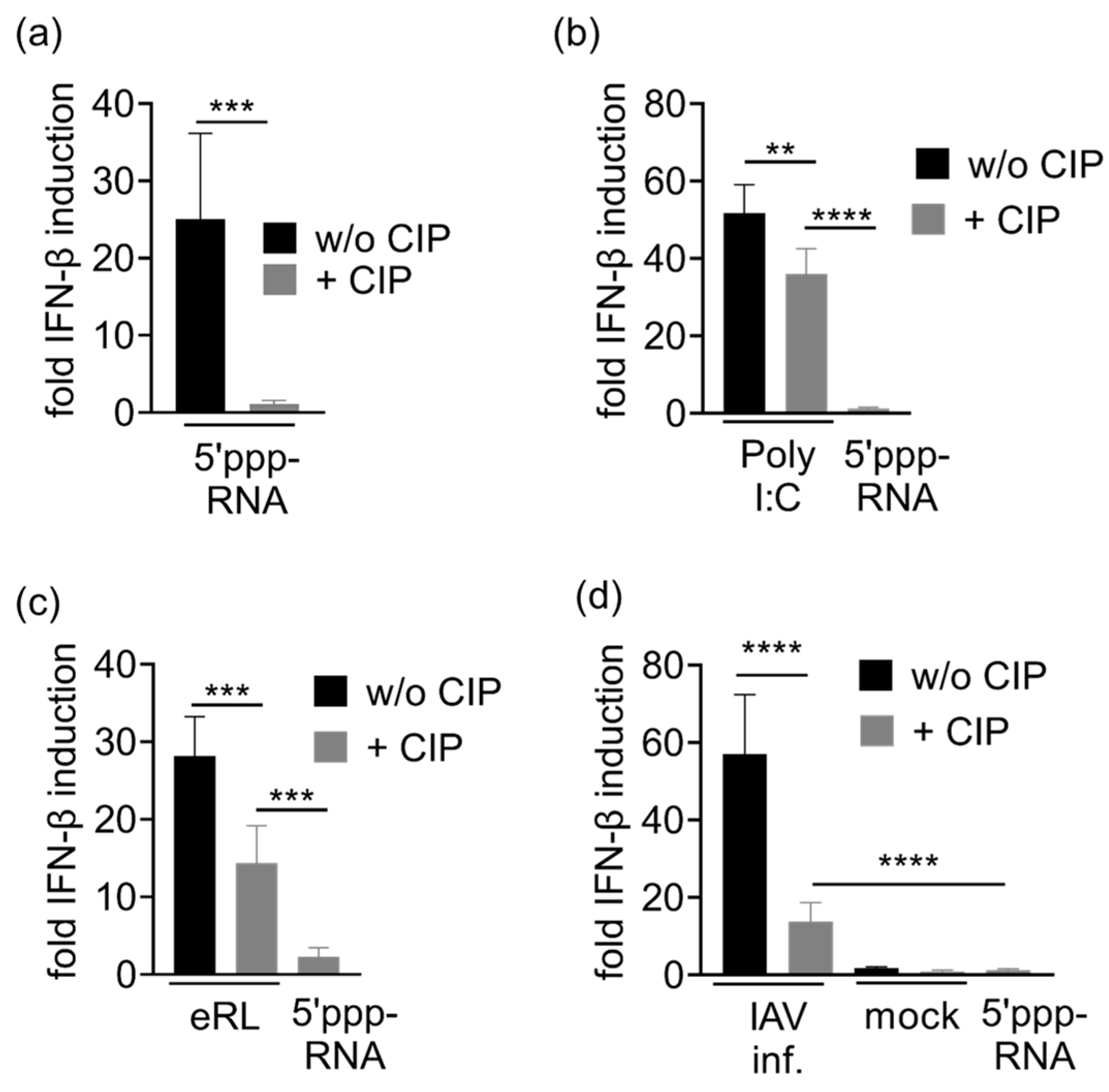
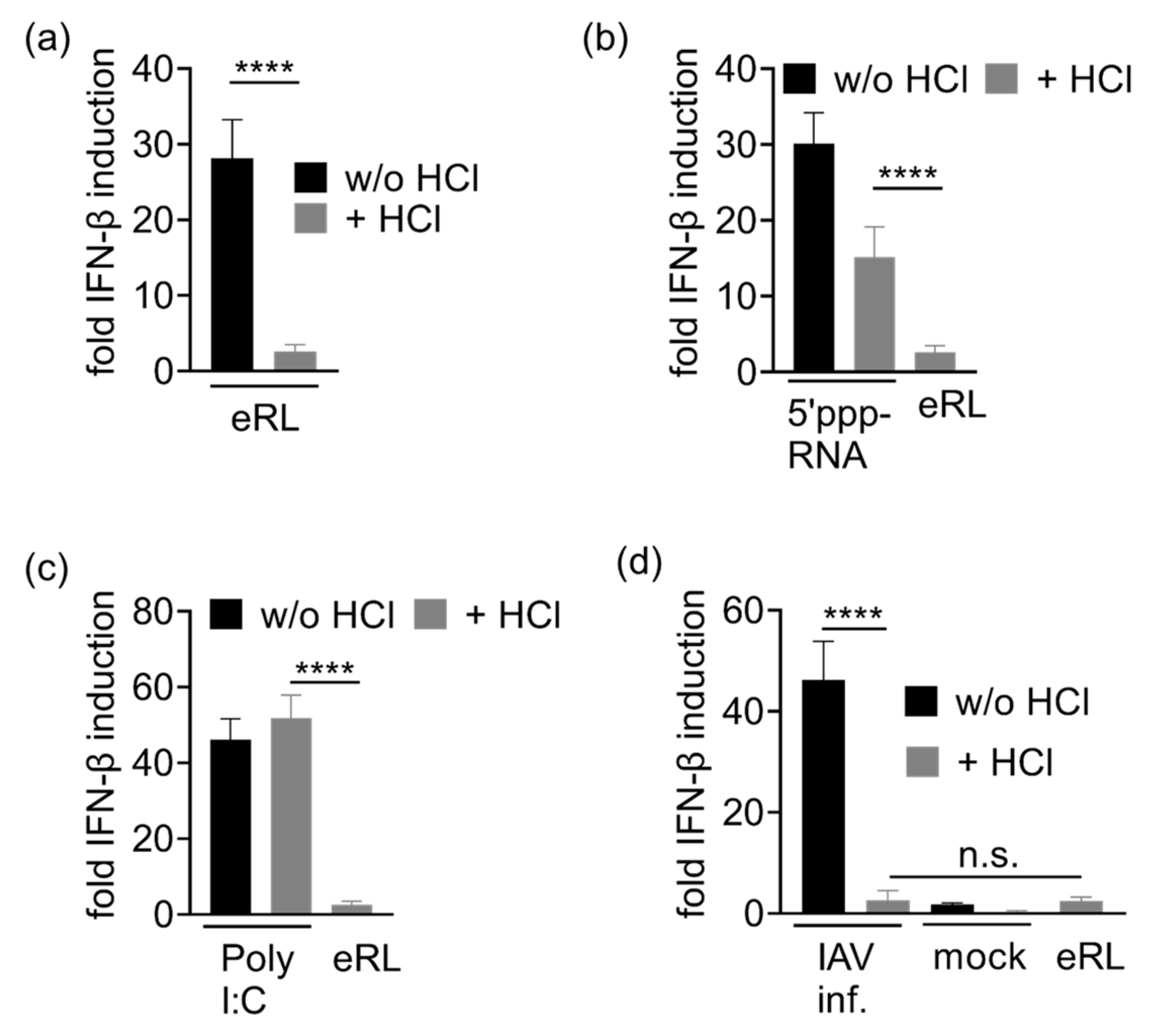
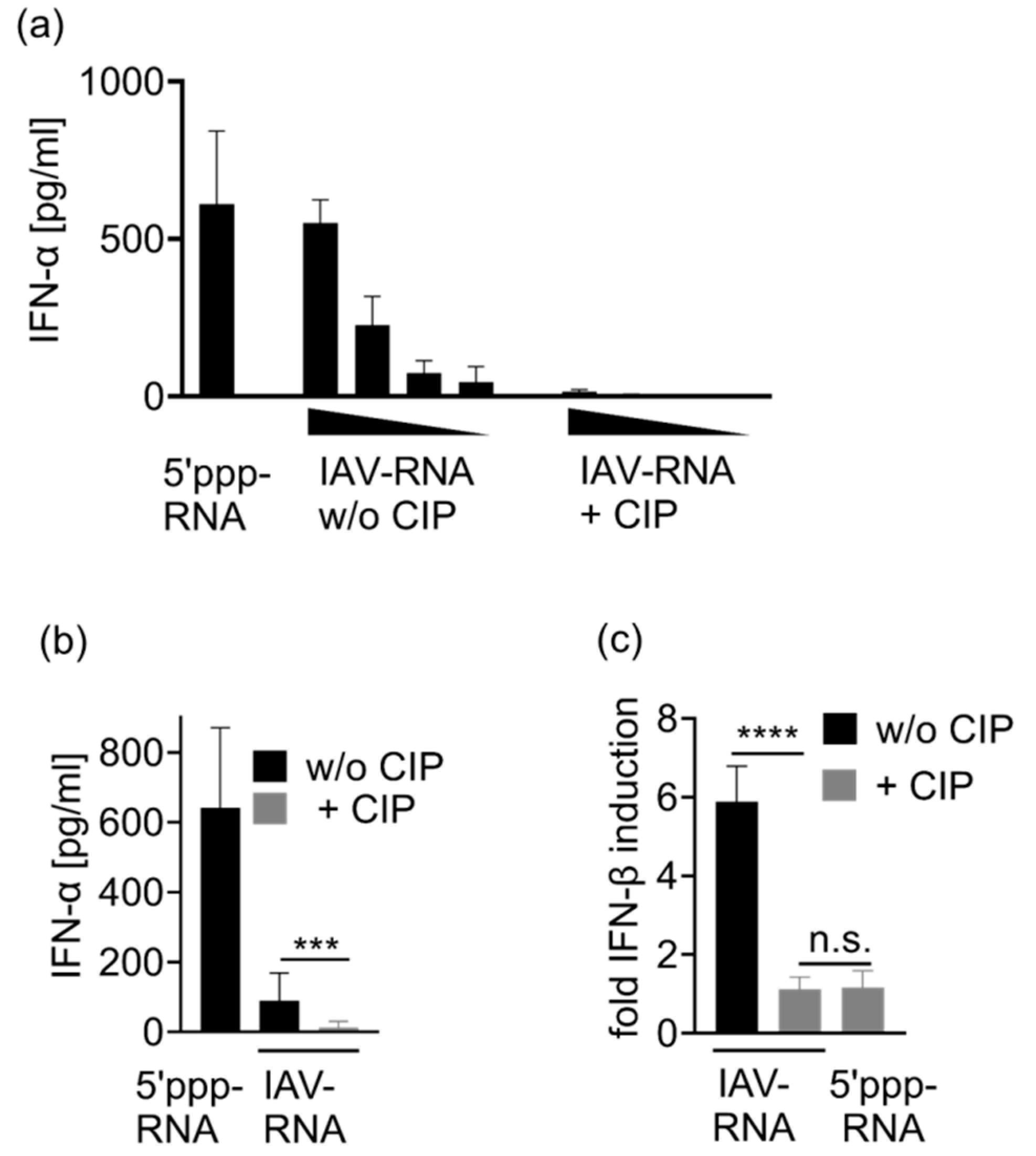
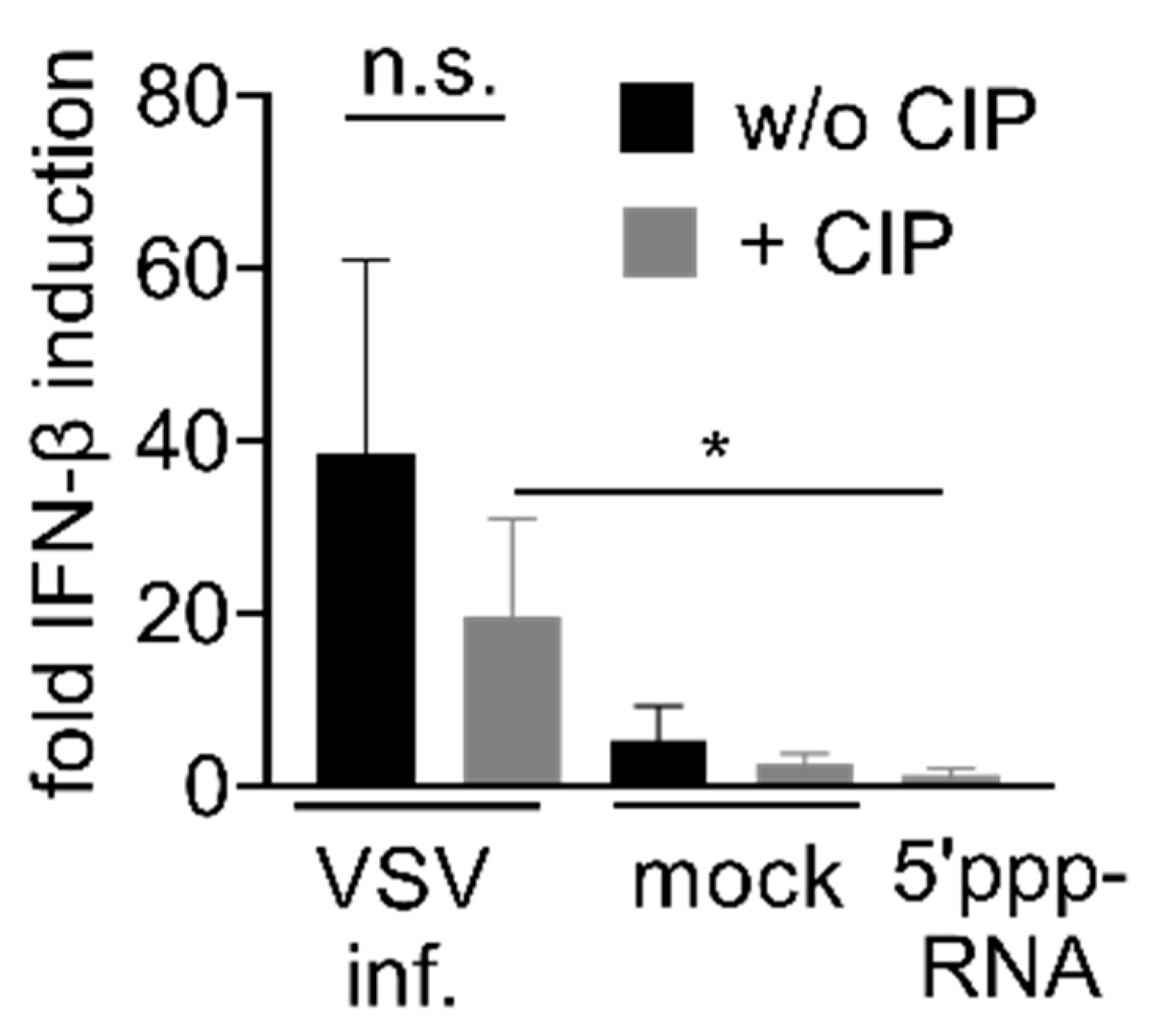
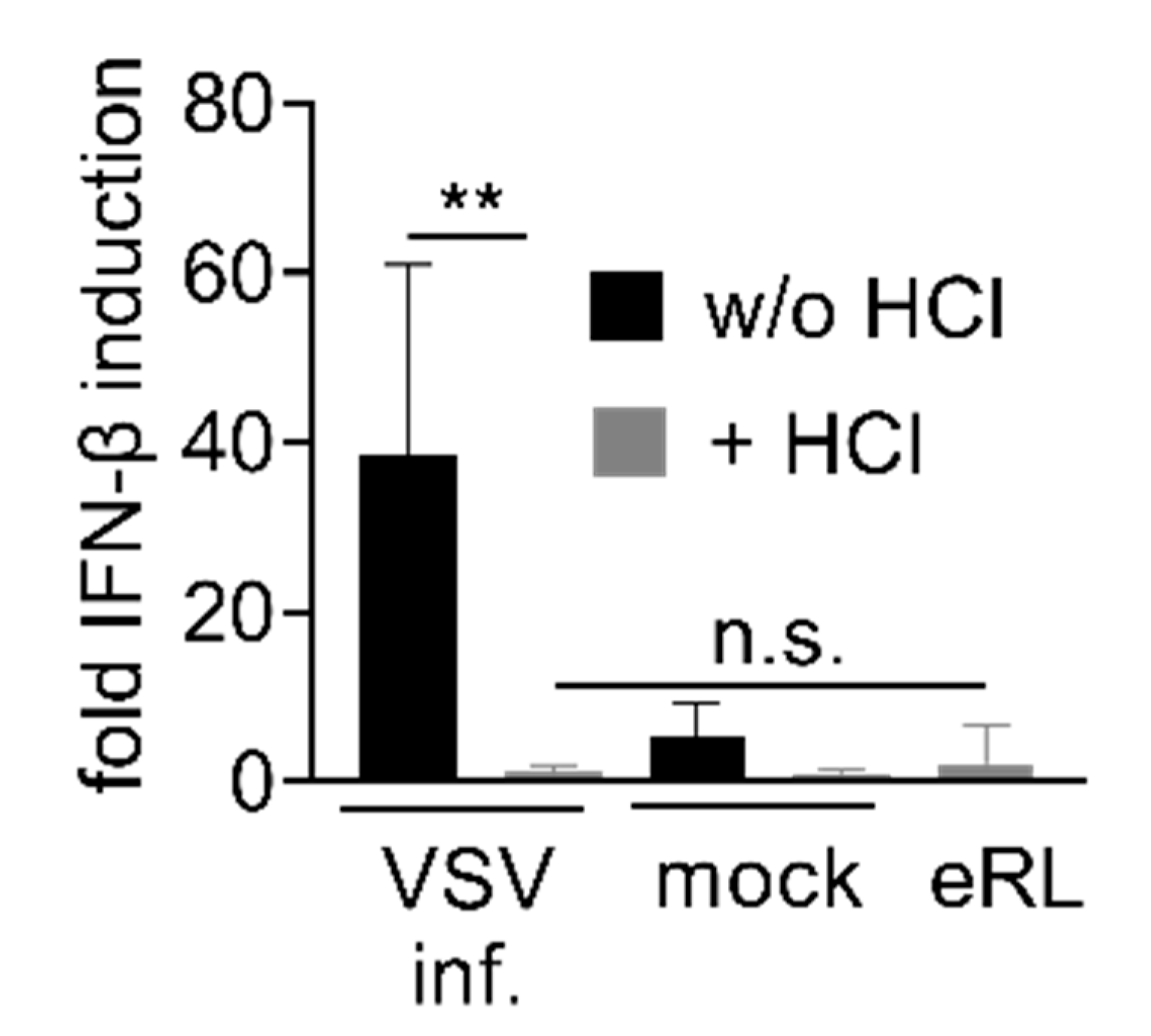
3.4. Immunostimulation by IAV-Induced RNA Depends on 2′3′ Cyclic Phosphate
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A






References
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef]
- Murphy, K.; Weaver, C. Janeway′s Immunobiology; Garland Science: Devon, UK, 2017. [Google Scholar]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, G. Nucleic acid immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5’-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; von Thülen, T.; Yang, I.; Laukemper, V.; Rupf, B.; Janga, H.; Panagiotidis, G.-D.; Schoen, A.; Nicolai, M.; Schulte, L.N.; et al. A ribosomal RNA fragment with 2′,3′-cyclic phosphate and GTP-binding activity acts as RIG-I ligand. Nucleic Acids Res. 2020, 48, 10397–10412. [Google Scholar] [CrossRef] [PubMed]
- Cauldwell, A.V.; Long, J.S.; Moncorgé, O.; Barclay, W.S. Viral determinants of influenza A virus host range. J. Gen. Virol. 2014, 95, 1193–1210. [Google Scholar] [CrossRef] [Green Version]
- Klenk, H.D. Influenza viruses en route from birds to man. Cell Host Microbe 2014, 15, 653–654. [Google Scholar] [CrossRef] [Green Version]
- Ampomah, P.B.; Lim, L.H.K. Influenza a virus-induced apoptosis and virus propagation. Apoptosis 2020, 25, 1–11. [Google Scholar] [CrossRef]
- Weber, M.; Gawanbacht, A.; Habjan, M.; Rang, A.; Borner, C.; Schmidt, A.M.; Veitinger, S.; Jacob, R.; Devignot, S.; Kochs, G.; et al. Incoming RNA virus nucleocapsids containing a 5’-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe 2013, 13, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.K.; Lopez-Martinez, A.; Altomonte, J. Oncolytic vesicular stomatitis virus as a viro-immunotherapy: Defeating cancer with a “Hammer” and “Anvil”. Biomedicines 2017, 5, 8. [Google Scholar] [CrossRef]
- Munis, A.M.; Bentley, E.M.; Takeuchi, Y. A tool with many applications: Vesicular stomatitis virus in research and medicine. Expert. Opin. Biol. Ther. 2020, 20, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Bishnoi, S.; Tiwari, R.; Gupta, S.; Byrareddy, S.N.; Nayak, D. Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in cancer therapy. Viruses 2018, 10, 90. [Google Scholar] [CrossRef] [Green Version]
- Brauer, R.; Chen, P. Influenza virus propagation in embryonated chicken eggs. J. Vis. Exp. 2015, 52421. [Google Scholar] [CrossRef] [Green Version]
- Honda, S.; Morichika, K.; Kirino, Y. Selective amplification and sequencing of cyclic phosphate-containing RNAs by the cP-RNA-seq method. Nat. Protoc. 2016, 11, 476–489. [Google Scholar] [CrossRef] [Green Version]
- Usher, D.A.; McHale, A.H. Hydrolytic stability of helical RNA: A selective advantage for the natural 3’,5’-bond. Proc. Natl. Acad. Sci. USA 1976, 73, 1149–1153. [Google Scholar] [CrossRef] [Green Version]
- Lutay, A.V.; Chernolovskaya, E.L.; Zenkova, M.A.; Vlassov, V.V. The nonenzymatic template-directed ligation of oligonucleotides. Biogeosciences 2006, 3, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Zenkova, M.A. Artificial Nucleases; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Eckard, S.C.; Rice, G.I.; Fabre, A.; Badens, C.; Gray, E.E.; Hartley, J.L.; Crow, Y.J.; Stetson, D.B. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat. Immunol. 2014, 15, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Gusho, E.; Baskar, D.; Banerjee, S. New advances in our understanding of the "unique" RNase L in host pathogen interaction and immune signaling. Cytokine 2020, 133, 153847. [Google Scholar] [CrossRef] [PubMed]
- Lassig, C.; Matheisl, S.; Sparrer, K.M.; de Oliveira Mann, C.C.; Moldt, M.; Patel, J.R.; Goldeck, M.; Hartmann, G.; Garcia-Sastre, A.; Hornung, V.; et al. ATP hydrolysis by the viral RNA sensor RIG-I prevents unintentional recognition of self-RNA. eLife 2015, 4. [Google Scholar] [CrossRef]
- Chiang, J.J.; Sparrer, K.M.J.; van Gent, M.; Lassig, C.; Huang, T.; Osterrieder, N.; Hopfner, K.P.; Gack, M.U. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 2018, 19, 53–62. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, S.; Yang, Z.; Lin, H.; Zhu, J.; Liu, L.; Wang, W.; Liu, S.; Liu, W.; Ma, Y.; et al. Self-recognition of an inducible host lncRNA by RIG-I feedback restricts innate immune response. Cell 2018, 173, 906–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2’-5’ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef] [Green Version]
- Behera, A.K.; Kumar, M.; Lockey, R.F.; Mohapatra, S.S. 2’-5’ Oligoadenylate synthetase plays a critical role in interferon-gamma inhibition of respiratory syncytial virus infection of human epithelial cells. J. Biol. Chem. 2002, 277, 25601–25608. [Google Scholar] [CrossRef] [Green Version]
- Malathi, K.; Saito, T.; Crochet, N.; Barton, D.J.; Gale, M., Jr.; Silverman, R.H. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 2010, 16, 2108–2119. [Google Scholar] [CrossRef] [Green Version]
- Greulich, W.; Wagner, M.; Gaidt, M.M.; Stafford, C.; Cheng, Y.; Linder, A.; Carell, T.; Hornung, V. TLR8 is a sensor of RNase T2 degradation products. Cell 2019, 179, 1264–1275.e1213. [Google Scholar] [CrossRef] [PubMed]
- Ostendorf, T.; Zillinger, T.; Andryka, K.; Schlee-Guimaraes, T.M.; Schmitz, S.; Marx, S.; Bayrak, K.; Linke, R.; Salgert, S.; Wegner, J.; et al. Immune sensing of synthetic, bacterial, and protozoan RNA by Toll-like receptor 8 requires coordinated processing by RNase T2 and RNase 2. Immunity 2020, 52, 591–605.e596. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinberg, J.; Wadenpohl, T.; Jung, S. The Endogenous RIG-I Ligand Is Generated in Influenza A-Virus Infected Cells. Viruses 2021, 13, 1564. https://doi.org/10.3390/v13081564
Steinberg J, Wadenpohl T, Jung S. The Endogenous RIG-I Ligand Is Generated in Influenza A-Virus Infected Cells. Viruses. 2021; 13(8):1564. https://doi.org/10.3390/v13081564
Chicago/Turabian StyleSteinberg, Julia, Timo Wadenpohl, and Stephanie Jung. 2021. "The Endogenous RIG-I Ligand Is Generated in Influenza A-Virus Infected Cells" Viruses 13, no. 8: 1564. https://doi.org/10.3390/v13081564
APA StyleSteinberg, J., Wadenpohl, T., & Jung, S. (2021). The Endogenous RIG-I Ligand Is Generated in Influenza A-Virus Infected Cells. Viruses, 13(8), 1564. https://doi.org/10.3390/v13081564