
Characterization of the SARS-CoV-2 Host Response in Primary Human Airway Epithelial Cells from Aged Individuals

 , and
, and 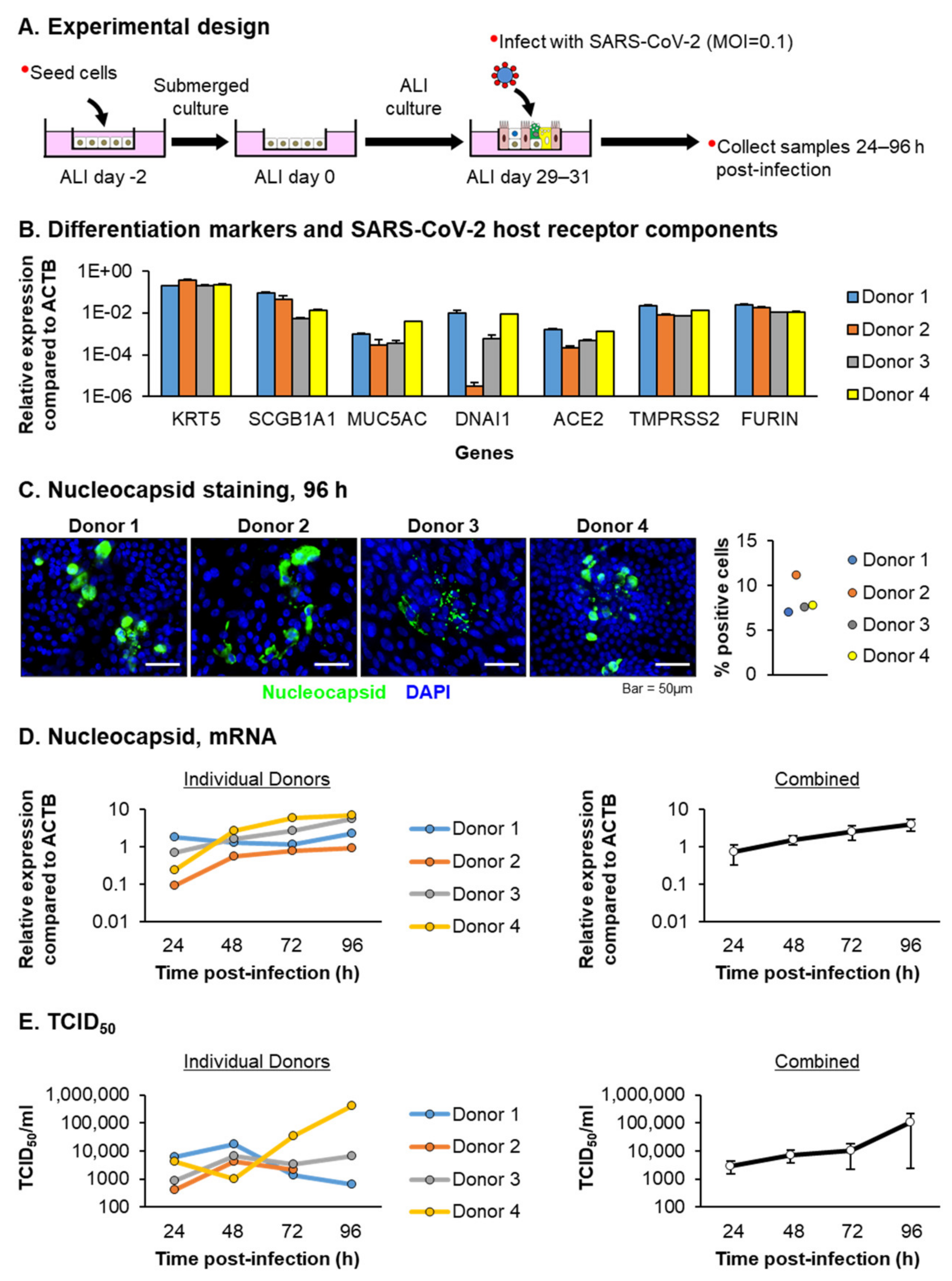{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation and Titration of Severe Acute Respiratory Syndrome-Related Coronavirus 2 (SARS-CoV-2) Stocks
2.2. Human Bronchial Epithelial Cell (HBEC) Cultures and Air–Liquid Interface (ALI) Culture
2.3. Infection of ALI Cultures
2.4. RNA Extraction and cDNA Synthesis
2.5. Gene Expression Analysis
2.6. Bulk RNA Sequencing (Bulk RNA-Seq)
2.7. Immunofluorescence Staining
2.8. Statistics
3. Results
3.1. HBECs from Aged Individuals Are Permissive to SARS-CoV-2 Infection
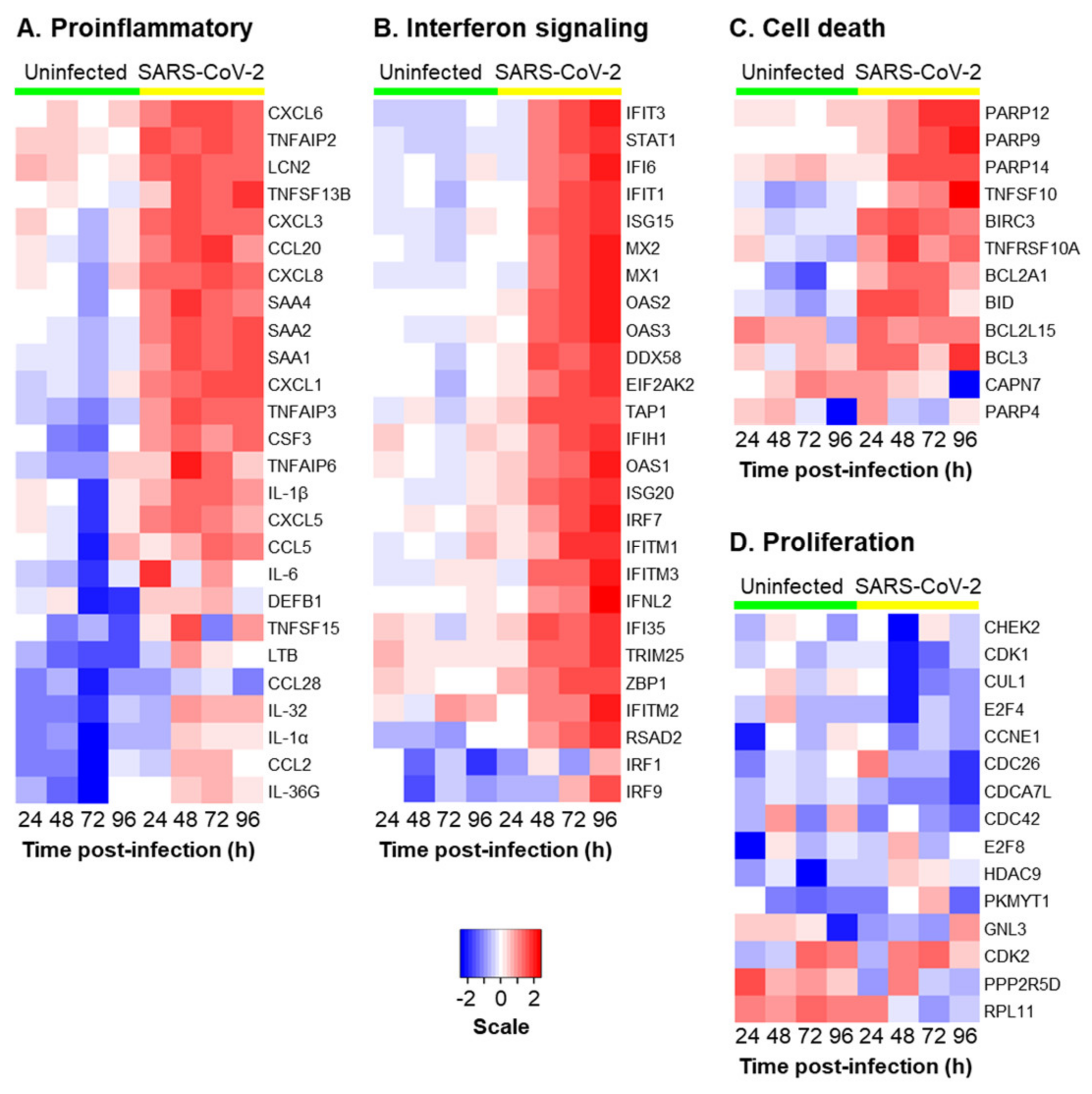
3.2. SARS-CoV-2 Infection Induces a Proinflammatory and Interferon Response in HBECs from Aged Individuals
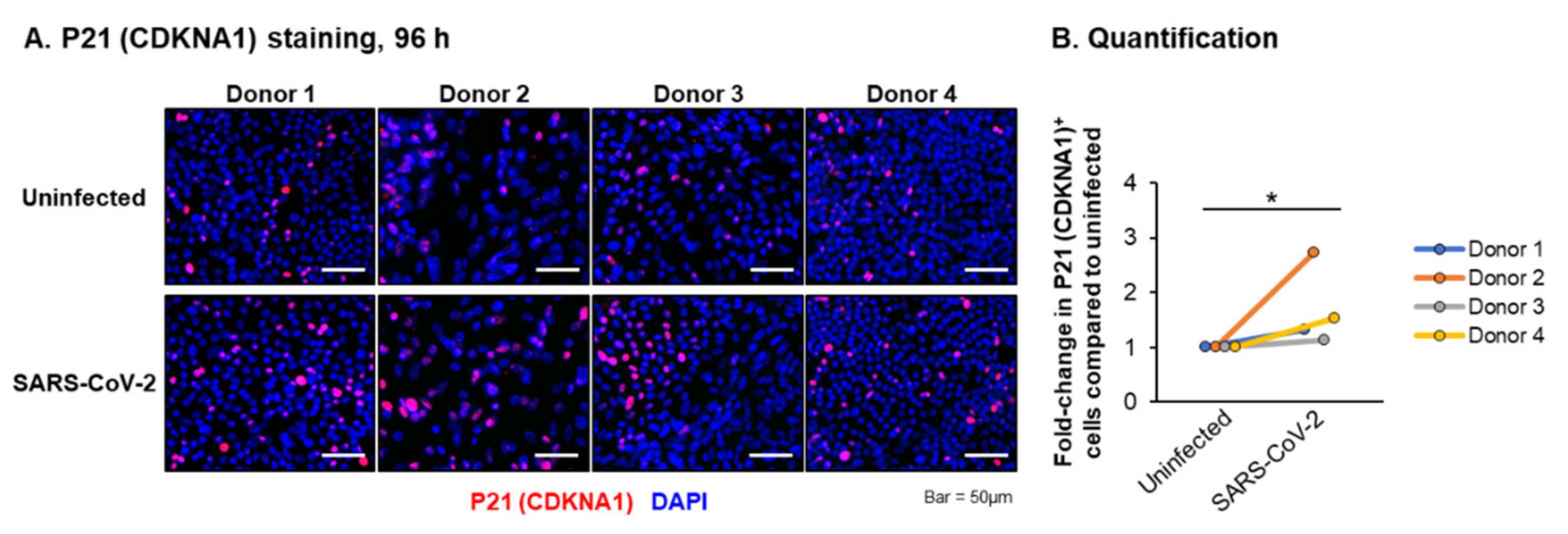
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 9 June 2021).
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 2020, 182, 429–446. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Cheung, M.C.; Perera, R.; Ng, K.C.; Bui, C.H.T.; Ho, J.C.W.; Ng, M.M.T.; Kuok, D.I.T.; Shih, K.C.; Tsao, S.W.; et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: An analysis in ex-vivo and in-vitro cultures. Lancet Respir. Med. 2020, 8, 687–695. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Rostami, M.R.; Leopold, P.L.; Mezey, J.G.; O’Beirne, S.L.; Strulovici-Barel, Y.; Crystal, R.G. expression of the sars-cov-2 ace2 receptor in the human airway epithelium. Am. J. Respir. Crit. Care Med. 2020, 202, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Heinen, N.; Klohn, M.; Steinmann, E.; Pfaender, S. In vitro lung models and their application to study SARS-CoV-2 Pathogenesis and disease. Viruses 2021, 13, 792. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced host response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef]
- Han, Y.; Duan, X.; Yang, L.; Nilsson-Payant, B.E.; Wang, P.; Duan, F.; Tang, X.; Yaron, T.M.; Zhang, T.; Uhl, S.; et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 2021, 589, 270–275. [Google Scholar] [CrossRef]
- Hao, S.; Ning, K.; Kuz, C.A.; Vorhies, K.; Yan, Z.; Qiu, J. Long-Term modeling of SARS-CoV-2 infection of In vitro cultured polarized human airway epithelium. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Katsura, H.; Sontake, V.; Tata, A.; Kobayashi, Y.; Edwards, C.E.; Heaton, B.E.; Konkimalla, A.; Asakura, T.; Mikami, Y.; Fritch, E.J.; et al. Human lung stem cell-based alveolospheres provide insights into SARS-CoV-2-mediated interferon responses and pneumocyte dysfunction. Cell Stem Cell 2020, 27, 890–904. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Mulay, A.; Konda, B.; Garcia, G., Jr.; Yao, C.; Beil, S.; Villalba, J.M.; Koziol, C.; Sen, C.; Purkayastha, A.; Kolls, J.K.; et al. SARS-CoV-2 infection of primary human lung epithelium for COVID-19 modeling and drug discovery. Cell Rep. 2021, 35, 109055. [Google Scholar] [CrossRef] [PubMed]
- Pruijssers, A.J.; George, A.S.; Schafer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H., 3rd; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir inhibits SARS-CoV-2 in human lung cells and chimeric SARS-CoV expressing the SARS-CoV-2 RNA polymerase in MICE. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, A.; Sen, C.; Garcia, G., Jr.; Langerman, J.; Shia, D.W.; Meneses, L.K.; Vijayaraj, P.; Durra, A.; Koloff, C.R.; Freund, D.R.; et al. Direct exposure to SARS-CoV-2 and cigarette smoke increases infection severity and alters the stem cell-derived airway repair response. Cell Stem Cell 2020, 27, 869–875. [Google Scholar] [CrossRef]
- Ravindra, N.G.; Alfajaro, M.M.; Gasque, V.; Huston, N.C.; Wan, H.; Szigeti-Buck, K.; Yasumoto, Y.; Greaney, A.M.; Habet, V.; Chow, R.D.; et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium identifies target cells, alterations in gene expression, and cell state changes. PLoS Biol. 2021, 19, e3001143. [Google Scholar] [CrossRef]
- Sajuthi, S.P.; DeFord, P.; Li, Y.; Jackson, N.D.; Montgomery, M.T.; Everman, J.L.; Rios, C.L.; Pruesse, E.; Nolin, J.D.; Plender, E.G.; et al. Type 2 and interferon inflammation regulate SARS-CoV-2 entry factor expression in the airway epithelium. Nat. Commun. 2020, 11, 5139. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Choi, S.S.; Rustagi, A.; Zhu, J.; van Unen, V.; de la, O.S.; Flynn, R.A.; Margalef-Catala, M.; Santos, A.J.M.; Ju, J.; et al. Progenitor identification and SARS-CoV-2 infection in human distal lung organoids. Nature 2020, 588, 670–675. [Google Scholar] [CrossRef]
- Si, L.; Bai, H.; Rodas, M.; Cao, W.; Oh, C.Y.; Jiang, A.; Moller, R.; Hoagland, D.; Oishi, K.; Horiuchi, S.; et al. A human-airway-on-a-chip for the rapid identification of candidate antiviral therapeutics and prophylactics. Nat. Biomed. Eng. 2021. [Google Scholar] [CrossRef] [PubMed]
- Vanderheiden, A.; Ralfs, P.; Chirkova, T.; Upadhyay, A.A.; Zimmerman, M.G.; Bedoya, S.; Aoued, H.; Tharp, G.M.; Pellegrini, K.L.; Manfredi, C.; et al. Type I and type III interferons restrict SARS-CoV-2 infection of human airway epithelial cultures. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- V’Kovski, P.; Gultom, M.; Kelly, J.N.; Steiner, S.; Russeil, J.; Mangeat, B.; Cora, E.; Pezoldt, J.; Holwerda, M.; Kratzel, A.; et al. Disparate temperature-dependent virus-host dynamics for SARS-CoV-2 and SARS-CoV in the human respiratory epithelium. PLoS Biol. 2021, 19, e3001158. [Google Scholar] [CrossRef]
- Youk, J.; Kim, T.; Evans, K.V.; Jeong, Y.I.; Hur, Y.; Hong, S.P.; Kim, J.H.; Yi, K.; Kim, S.Y.; Na, K.J.; et al. Three-dimensional human alveolar stem cell culture models reveal infection response to SARS-CoV-2. Cell Stem Cell 2020, 27, 905–919. [Google Scholar] [CrossRef]
- Zhu, N.; Wang, W.; Liu, Z.; Liang, C.; Wang, W.; Ye, F.; Huang, B.; Zhao, L.; Wang, H.; Zhou, W.; et al. Morphogenesis and cytopathic effect of SARS-CoV-2 infection in human airway epithelial cells. Nat. Commun. 2020, 11, 3910. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pohlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 2020, 78, 779–784.e775. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020, 181, 1016–1035.e1019. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, A.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021. [Google Scholar] [CrossRef]
- Hue, S.; Beldi-Ferchiou, A.; Bendib, I.; Surenaud, M.; Fourati, S.; Frapard, T.; Rivoal, S.; Razazi, K.; Carteaux, G.; Delfau-Larue, M.H.; et al. Uncontrolled innate and impaired adaptive immune responses in patients with COVID-19 acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2020, 202, 1509–1519. [Google Scholar] [CrossRef]
- Mangalmurti, N.; Hunter, C.A. Cytokine Storms: Understanding COVID-19. Immunity 2020, 53, 19–25. [Google Scholar] [CrossRef] [PubMed]
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Ni Choileain, O.; Clarke, J.; O’Connor, E.; Hogan, G.; et al. Characterization of the inflammatory response to severe COVID-19 illness. Am. J. Respir. Crit. Care Med. 2020, 202, 812–821. [Google Scholar] [CrossRef]
- Mick, E.; Kamm, J.; Pisco, A.O.; Ratnasiri, K.; Babik, J.M.; Castaneda, G.; DeRisi, J.L.; Detweiler, A.M.; Hao, S.L.; Kangelaris, K.N.; et al. Upper airway gene expression reveals suppressed immune responses to SARS-CoV-2 compared with other respiratory viruses. Nat. Commun. 2020, 11, 5854. [Google Scholar] [CrossRef]
- Zhang, F.; Gan, R.; Zhen, Z.; Hu, X.; Li, X.; Zhou, F.; Liu, Y.; Chen, C.; Xie, S.; Zhang, B.; et al. Adaptive immune responses to SARS-CoV-2 infection in severe versus mild individuals. Signal Transduct. Target. Ther. 2020, 5, 156. [Google Scholar] [CrossRef]
- Gasmi, A.; Peana, M.; Pivina, L.; Srinath, S.; Gasmi Benahmed, A.; Semenova, Y.; Menzel, A.; Dadar, M.; Bjorklund, G. Interrelations between COVID-19 and other disorders. Clin. Immunol. 2021, 224, 108651. [Google Scholar] [CrossRef]
- Lowe, K.E.; Zein, J.; Hatipoglu, U.; Attaway, A. Association of Smoking and Cumulative Pack-Year Exposure With COVID-19 Outcomes in the Cleveland Clinic COVID-19 Registry. JAMA Intern. Med. 2021, 181, 709–711. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F. Aging, male sex, obesity, and metabolic inflammation create the perfect storm for COVID-19. Diabetes 2020, 69, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.H.; Tipih, T.; Makoah, N.A.; Vermeulen, J.G.; Goedhals, D.; Sempa, J.B.; Burt, F.J.; Taylor, A.; Mahalingam, S. Comorbidities in SARS-CoV-2 patients: A systematic review and meta-analysis. mBio 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, immunity, and COVID-19: How age influences the host immune response to coronavirus infections? Front. Physiol. 2020, 11, 571416. [Google Scholar] [CrossRef]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2021, 65, 101205. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 1938, 27, 493–497. [Google Scholar]
- Bodas, M.; Moore, A.R.; Subramaniyan, B.; Georgescu, C.; Wren, J.D.; Freeman, W.M.; Brown, B.R.; Metcalf, J.P.; Walters, M.S. Cigarette smoke activates NOTCH3 to promote goblet cell differentiation in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2021, 64, 426–440. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, G.E.; Roussos, P. Dream: Powerful differential expression analysis for repeated measures designs. Bioinformatics 2021, 37, 192–201. [Google Scholar] [CrossRef]
- Chlamydas, S.; Papavassiliou, A.G.; Piperi, C. Epigenetic mechanisms regulating COVID-19 infection. Epigenetics 2021, 16, 263–270. [Google Scholar] [CrossRef]
- Forcados, G.E.; Muhammad, A.; Oladipo, O.O.; Makama, S.; Meseko, C.A. Metabolic Implications of Oxidative Stress and Inflammatory Process in Sars-Cov-2 Pathogenesis: Therapeutic Potential of Natural Antioxidants. Front. Cell. Infect. Microbiol. 2021, 11, 654813. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Garbati, M.; Bryant, K.; Lu, Y. Epigenetic mechanisms influencing COVID-19. Genome 2021, 64, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of oxidative stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) infection: A review. Protein. J. 2020, 39, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 2021. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Cellular senescence and lung function during aging. Yin and Yang. Ann. Am. Thorac. Soc. 2016, 13, S402–S406. [Google Scholar] [CrossRef] [Green Version]
- Hamsanathan, S.; Alder, J.K.; Sellares, J.; Rojas, M.; Gurkar, A.U.; Mora, A.L. Cellular senescence: The trojan horse in chronic lung diseases. Am. J. Respir. Cell. Mol. Biol. 2019, 61, 21–30. [Google Scholar] [CrossRef]
- Parikh, P.; Wicher, S.; Khandalavala, K.; Pabelick, C.M.; Britt, R.D., Jr.; Prakash, Y.S. Cellular senescence in the lung across the age spectrum. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L826–L842. [Google Scholar] [CrossRef]
- Kelley, W.J.; Zemans, R.L.; Goldstein, D.R. Cellular senescence: Friend or foe to respiratory viral infections? Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef] [PubMed]
- Seoane, R.; Vidal, S.; Bouzaher, Y.H.; El Motiam, A.; Rivas, C. The interaction of viruses with the cellular senescence response. Biology 2020, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Nehme, J.; Borghesan, M.; Mackedenski, S.; Bird, T.G.; Demaria, M. Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell 2020, 19, e13237. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramaniyan, B.; Larabee, J.L.; Bodas, M.; Moore, A.R.; Burgett, A.W.G.; Myers, D.A.; Georgescu, C.; Wren, J.D.; Papin, J.F.; Walters, M.S. Characterization of the SARS-CoV-2 Host Response in Primary Human Airway Epithelial Cells from Aged Individuals. Viruses 2021, 13, 1603. https://doi.org/10.3390/v13081603
Subramaniyan B, Larabee JL, Bodas M, Moore AR, Burgett AWG, Myers DA, Georgescu C, Wren JD, Papin JF, Walters MS. Characterization of the SARS-CoV-2 Host Response in Primary Human Airway Epithelial Cells from Aged Individuals. Viruses. 2021; 13(8):1603. https://doi.org/10.3390/v13081603
Chicago/Turabian StyleSubramaniyan, Bharathiraja, Jason L. Larabee, Manish Bodas, Andrew R. Moore, Anthony W. G. Burgett, Dean A. Myers, Constantin Georgescu, Jonathan D. Wren, James F. Papin, and Matthew S. Walters. 2021. "Characterization of the SARS-CoV-2 Host Response in Primary Human Airway Epithelial Cells from Aged Individuals" Viruses 13, no. 8: 1603. https://doi.org/10.3390/v13081603
APA StyleSubramaniyan, B., Larabee, J. L., Bodas, M., Moore, A. R., Burgett, A. W. G., Myers, D. A., Georgescu, C., Wren, J. D., Papin, J. F., & Walters, M. S. (2021). Characterization of the SARS-CoV-2 Host Response in Primary Human Airway Epithelial Cells from Aged Individuals. Viruses, 13(8), 1603. https://doi.org/10.3390/v13081603