

EBV Association with Lymphomas and Carcinomas in the Oral Compartment


Abstract
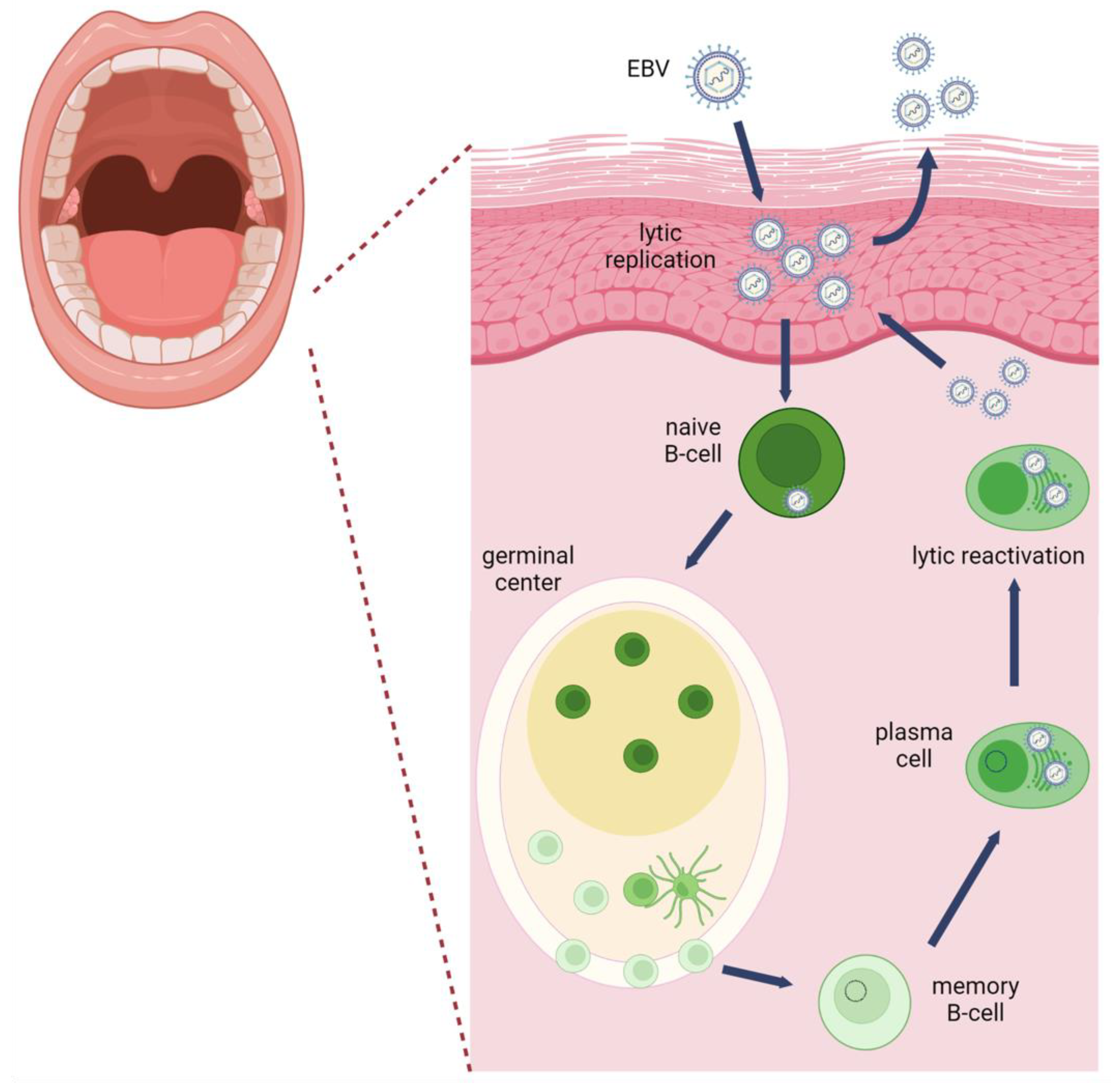
:1. Introduction
2. Mechanisms of EBV Oncogenesis
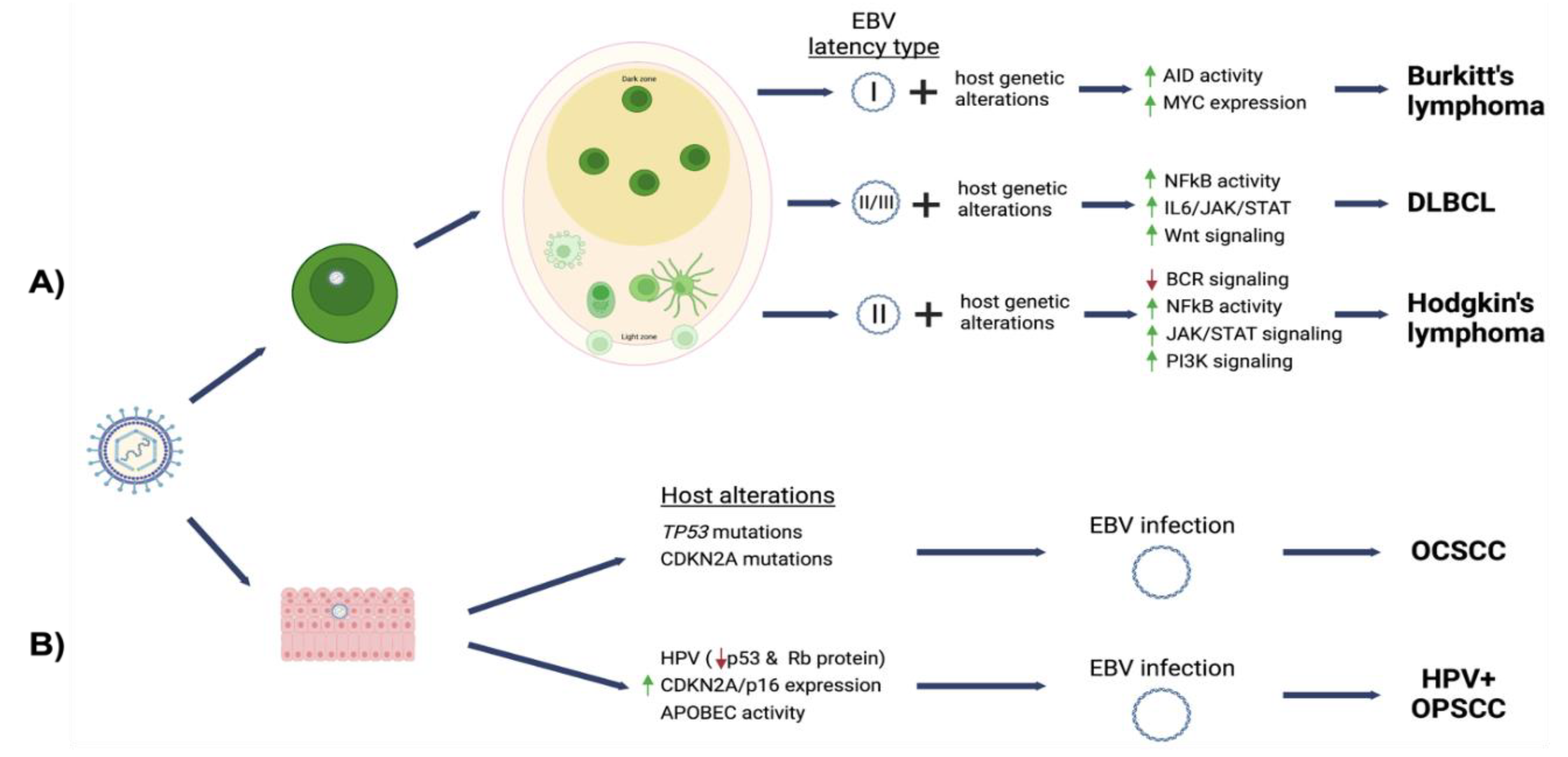
3. EBV-Associated Oral Lymphoma
3.1. Burkitt’s Lymphoma
3.2. Diffuse Large B Cell Lymphoma
3.3. Hodgkin Lymphoma
4. EBV-Associated Oral Carcinomas
4.1. Oral Squamous Cell Carcinoma
4.2. HPV-Positive Oropharyngeal Squamous Cell Carcinoma
4.3. Oral Hairy Leukoplakia
4.4. Lymphoepitheliomas of the Salivary/Parotid Gland
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV persistence in memory B cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sixbey, J.W.; Vesterinen, E.H.; Nedrud, J.G.; Raab-Traub, N.; Walton, L.A.; Pagano, J.S. Replication of Epstein-Barr virus in human epithelial cells infected in vitro. Nature 1983, 306, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Hutt-Fletcher, L.M. The Long and Complicated Relationship between Epstein-Barr Virus and Epithelial Cells. J. Virol. 2017, 91, e01677-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawandar, D.M.; Ohashi, M.; Djavadian, R.; Barlow, E.; Makielski, K.; Ali, A.; Lee, D.; Lambert, P.F.; Johannsen, E.; Kenney, S.C. Differentiation-Dependent LMP1 Expression Is Required for Efficient Lytic Epstein-Barr Virus Reactivation in Epithelial Cells. J. Virol. 2017, 91, e02438-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henle, G.; Henle, W. Observations on childhood infections with the Epstein-Barr virus. J. Infect. Dis. 1970, 121, 303–310. [Google Scholar] [CrossRef]
- Niederman, J.C.; Miller, G.; Pearson, H.A.; Pagano, J.S.; Dowaliby, J.M. Infectious mononucleosis. Epstein-Barr-virus shedding in saliva and the oropharynx. N. Engl. J. Med. 1976, 294, 1355–1359. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.Y.; Rickinson, A.B.; Epstein, M.A. A re-examination of the Epstein-Barr virus carrier state in healthy seropositive individuals. Int. J. Cancer 1985, 35, 35–42. [Google Scholar] [CrossRef]
- Hadinoto, V.; Shapiro, M.; Sun, C.C.; Thorley-Lawson, D.A. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 2009, 5, e1000496. [Google Scholar] [CrossRef] [Green Version]
- Fafi-Kremer, S.; Morand, P.; Brion, J.P.; Pavese, P.; Baccard, M.; Germi, R.; Genoulaz, O.; Nicod, S.; Jolivet, M.; Ruigrok, R.W.; et al. Long-term shedding of infectious epstein-barr virus after infectious mononucleosis. J. Infect. Dis. 2005, 191, 985–989. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.S.; Berger, J.R.; Mootoor, Y.; Avdiushko, S.A.; Zhu, H.; Kryscio, R.J. High prevalence of multiple human herpesviruses in saliva from human immunodeficiency virus-infected persons in the era of highly active antiretroviral therapy. J. Clin. Microbiol. 2006, 44, 2409–2415. [Google Scholar] [CrossRef]
- Shmakova, A.; Germini, D.; Vassetzky, Y. HIV-1, HAART and cancer: A complex relationship. Int. J. Cancer 2020, 146, 2666–2679. [Google Scholar] [CrossRef]
- Vincent-Bugnas, S.; Vitale, S.; Mouline, C.C.; Khaali, W.; Charbit, Y.; Mahler, P.; Prêcheur, I.; Hofman, P.; Maryanski, J.L.; Doglio, A. EBV infection is common in gingival epithelial cells of the periodontium and worsens during chronic periodontitis. PLoS ONE 2013, 8, e80336. [Google Scholar] [CrossRef] [Green Version]
- Tugizov, S.M.; Herrera, R.; Palefsky, J.M. Epstein-Barr virus transcytosis through polarized oral epithelial cells. J. Virol. 2013, 87, 8179–8194. [Google Scholar] [CrossRef] [Green Version]
- Miller, N.; Hutt-Fletcher, L.M. Epstein-Barr virus enters B cells and epithelial cells by different routes. J. Virol. 1992, 66, 3409–3414. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; Perez White, B.E.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef]
- Zimmermann, J.; Hammerschmidt, W. Structure and role of the terminal repeats of Epstein-Barr virus in processing and packaging of virion DNA. J. Virol. 1995, 69, 3147–3155. [Google Scholar] [CrossRef] [Green Version]
- Borza, C.M.; Hutt-Fletcher, L.M. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 2002, 8, 594–599. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Adland, E.; Bell, A.I.; Delecluse, H.J.; Rickinson, A.B.; Rowe, M. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: Viral genome expression, genome maintenance, and genome amplification. J. Virol. 2009, 83, 7749–7760. [Google Scholar] [CrossRef] [Green Version]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.; Takada, K.; Lui, V.W.; Lung, M.L.; et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef] [Green Version]
- Temple, R.M.; Zhu, J.; Budgeon, L.; Christensen, N.D.; Meyers, C.; Sample, C.E. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc. Natl. Acad. Sci. USA 2014, 111, 16544–16549. [Google Scholar] [CrossRef]
- Nawandar, D.M.; Wang, A.; Makielski, K.; Lee, D.; Ma, S.; Barlow, E.; Reusch, J.; Jiang, R.; Wille, C.K.; Greenspan, D.; et al. Differentiation-Dependent KLF4 Expression Promotes Lytic Epstein-Barr Virus Infection in Epithelial Cells. PLoS Pathog. 2015, 11, e1005195. [Google Scholar] [CrossRef] [PubMed]
- Kurth, J.; Hansmann, M.L.; Rajewsky, K.; Küppers, R. Epstein-Barr virus-infected B cells expanding in germinal centers of infectious mononucleosis patients do not participate in the germinal center reaction. Proc. Natl. Acad. Sci. USA 2003, 100, 4730–4735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fingeroth, J.D.; Weis, J.J.; Tedder, T.F.; Strominger, J.L.; Biro, P.A.; Fearon, D.T. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc. Natl. Acad. Sci. USA 1984, 81, 4510–4514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, T.; Adams, A.; Bjursell, G.; Bornkamm, G.W.; Kaschka-Dierich, C.; Jehn, U. Covalently closed circular duplex DNA of Epstein-Barr virus in a human lymphoid cell line. J. Mol. Biol. 1976, 102, 511–530. [Google Scholar] [CrossRef] [PubMed]
- Shirakata, M.; Imadome, K.I.; Hirai, K. Requirement of replication licensing for the dyad symmetry element-dependent replication of the Epstein-Barr virus oriP minichromosome. Virology 1999, 263, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Price, A.M.; Tourigny, J.P.; Forte, E.; Salinas, R.E.; Dave, S.S.; Luftig, M.A. Analysis of Epstein-Barr virus-regulated host gene expression changes through primary B-cell outgrowth reveals delayed kinetics of latent membrane protein 1-mediated NF-κB activation. J. Virol. 2012, 86, 11096–11106. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Kondo, Y.; Sugimoto, A.; Kawashima, D.; Saito, S.; Isomura, H.; Kanda, T.; Tsurumi, T. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in Raji cells. J. Virol. 2012, 86, 4752–4761. [Google Scholar] [CrossRef] [Green Version]
- Babcock, G.J.; Thorley-Lawson, D.A. Tonsillar memory B cells, latently infected with Epstein-Barr virus, express the restricted pattern of latent genes previously found only in Epstein-Barr virus-associated tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 12250–12255. [Google Scholar] [CrossRef] [Green Version]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Raver, R.M.; Panfil, A.R.; Hagemeier, S.R.; Kenney, S.C. The B-cell-specific transcription factor and master regulator Pax5 promotes Epstein-Barr virus latency by negatively regulating the viral immediate early protein BZLF1. J. Virol. 2013, 87, 8053–8063. [Google Scholar] [CrossRef]
- Bakkalci, D.; Jia, Y.; Winter, J.R.; Lewis, J.E.; Taylor, G.S.; Stagg, H.R. Risk factors for Epstein Barr virus-associated cancers: A systematic review, critical appraisal, and mapping of the epidemiological evidence. J. Glob. Health 2020, 10, 010405. [Google Scholar] [CrossRef]
- Pattengale, P.K.; Smith, R.W.; Gerber, P. Selective transformation of B lymphocytes by E.B. virus. Lancet 1973, 2, 93–94. [Google Scholar] [CrossRef]
- Thomas, J.A.; Allday, M.J.; Crawford, D.H. Epstein-Barr virus-associated lymphoproliferative disorders in immunocompromised individuals. Adv. Cancer Res. 1991, 57, 329–380. [Google Scholar] [CrossRef]
- Birdwell, C.E.; Prasai, K.; Dykes, S.; Jia, Y.; Munroe, T.G.C.; Bienkowska-Haba, M.; Scott, R.S. Epstein-Barr virus stably confers an invasive phenotype to epithelial cells through reprogramming of the WNT pathway. Oncotarget 2018, 9, 10417–10435. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [Green Version]
- Yee, J.; White, R.E.; Anderton, E.; Allday, M.J. Latent Epstein-Barr virus can inhibit apoptosis in B cells by blocking the induction of NOXA expression. PLoS ONE 2011, 6, e28506. [Google Scholar] [CrossRef] [Green Version]
- Hjalgrim, H.; Askling, J.; Sørensen, P.; Madsen, M.; Rosdahl, N.; Storm, H.H.; Hamilton-Dutoit, S.; Eriksen, L.S.; Frisch, M.; Ekbom, A.; et al. Risk of Hodgkin’s disease and other cancers after infectious mononucleosis. J. Natl. Cancer Inst. 2000, 92, 1522–1528. [Google Scholar] [CrossRef] [Green Version]
- Geser, A.; de Thé, G.; Lenoir, G.; Day, N.E.; Williams, E.H. Final case reporting from the Ugandan prospective study of the relationship between EBV and Burkitt’s lymphoma. Int. J. Cancer 1982, 29, 397–400. [Google Scholar] [CrossRef]
- De-Thé, G.; Geser, A.; Day, N.E.; Tukei, P.M.; Williams, E.H.; Beri, D.P.; Smith, P.G.; Dean, A.G.; Bronkamm, G.W.; Feorino, P.; et al. Epidemiological evidence for causal relationship between Epstein-Barr virus and Burkitt’s lymphoma from Ugandan prospective study. Nature 1978, 274, 756–761. [Google Scholar] [CrossRef]
- Kamranvar, S.A.; Gruhne, B.; Szeles, A.; Masucci, M.G. Epstein-Barr virus promotes genomic instability in Burkitt’s lymphoma. Oncogene 2007, 26, 5115–5123. [Google Scholar] [CrossRef]
- Gruhne, B.; Sompallae, R.; Masucci, M.G. Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 2009, 28, 3997–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epeldegui, M.; Hung, Y.P.; McQuay, A.; Ambinder, R.F.; Martínez-Maza, O. Infection of human B cells with Epstein-Barr virus results in the expression of somatic hypermutation-inducing molecules and in the accrual of oncogene mutations. Mol. Immunol. 2007, 44, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, M.M.L.; Lung, M.L. The Impact of Epstein-Barr Virus Infection on Epigenetic Regulation of Host Cell Gene Expression in Epithelial and Lymphocytic Malignancies. Front. Oncol. 2021, 11, 629780. [Google Scholar] [CrossRef] [PubMed]
- Usui, G.; Matsusaka, K.; Mano, Y.; Urabe, M.; Funata, S.; Fukayama, M.; Ushiku, T.; Kaneda, A. DNA Methylation and Genetic Aberrations in Gastric Cancer. Digestion 2021, 102, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Allday, M.J.; Bazot, Q.; White, R.E. The EBNA3 Family: Two Oncoproteins and a Tumour Suppressor that Are Central to the Biology of EBV in B Cells. Curr. Top. Microbiol. Immunol. 2015, 391, 61–117. [Google Scholar] [CrossRef]
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Jha, H.C.; Upadhyay, S.K.; Robertson, E.S. Epigenetic silencing of tumor suppressor genes during in vitro Epstein-Barr virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E5199–E5207. [Google Scholar] [CrossRef] [Green Version]
- Hernando, H.; Shannon-Lowe, C.; Islam, A.B.; Al-Shahrour, F.; Rodríguez-Ubreva, J.; Rodríguez-Cortez, V.C.; Javierre, B.M.; Mangas, C.; Fernández, A.F.; Parra, M.; et al. The B cell transcription program mediates hypomethylation and overexpression of key genes in Epstein-Barr virus-associated proliferative conversion. Genome Biol. 2013, 14, R3. [Google Scholar] [CrossRef] [Green Version]
- Mawardi, H.; Cutler, C.; Treister, N. Medical management update: Non-Hodgkin lymphoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontology 2009, 107, e19–e33. [Google Scholar] [CrossRef]
- Silva, T.D.; Ferreira, C.B.; Leite, G.B.; de Menezes Pontes, J.R.; Antunes, H.S. Oral manifestations of lymphoma: A systematic review. Ecancermedicalscience 2016, 10, 665. [Google Scholar] [CrossRef] [Green Version]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Burkitt’s Lymphoma. N. Engl. J. Med. 2022, 387, 1111–1122. [Google Scholar] [CrossRef]
- Burkitt, D.P. Etiology of Burkitt’s lymphoma--an alternative hypothesis to a vectored virus. J. Natl. Cancer Inst. 1969, 42, 19–28. [Google Scholar]
- Hämmerl, L.; Colombet, M.; Rochford, R.; Ogwang, D.M.; Parkin, D.M. The burden of Burkitt lymphoma in Africa. Infect. Agents Cancer 2019, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef] [Green Version]
- Bornkamm, G.W. Epstein-Barr virus and the pathogenesis of Burkitt’s lymphoma: More questions than answers. Int. J. Cancer 2009, 124, 1745–1755. [Google Scholar] [CrossRef]
- Lenze, D.; Leoncini, L.; Hummel, M.; Volinia, S.; Liu, C.G.; Amato, T.; De Falco, G.; Githanga, J.; Horn, H.; Nyagol, J.; et al. The different epidemiologic subtypes of Burkitt lymphoma share a homogenous micro RNA profile distinct from diffuse large B-cell lymphoma. Leukemia 2011, 25, 1869–1876. [Google Scholar] [CrossRef] [Green Version]
- Blum, K.A.; Lozanski, G.; Byrd, J.C. Adult Burkitt leukemia and lymphoma. Blood 2004, 104, 3009–3020. [Google Scholar] [CrossRef]
- Ziegler, J.L. Burkitt’s lymphoma. N. Engl. J. Med. 1981, 305, 735–745. [Google Scholar] [CrossRef]
- Queiroga, E.M.; Gualco, G.; Weiss, L.M.; Dittmer, D.P.; Araujo, I.; Klumb, C.E.; Harrington, W.J., Jr.; Bacchi, C.E. Burkitt lymphoma in Brazil is characterized by geographically distinct clinicopathologic features. Am. J. Clin. Pathol. 2008, 130, 946–956. [Google Scholar] [CrossRef] [Green Version]
- Sabattini, E.; Bacci, F.; Sagramoso, C.; Pileri, S.A. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: An overview. Pathologica 2010, 102, 83–87. [Google Scholar] [PubMed]
- Lim, S.T.; Karim, R.; Nathwani, B.N.; Tulpule, A.; Espina, B.; Levine, A.M. AIDS-related Burkitt’s lymphoma versus diffuse large-cell lymphoma in the pre-highly active antiretroviral therapy (HAART) and HAART eras: Significant differences in survival with standard chemotherapy. J. Clin. Oncol. 2005, 23, 4430–4438. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Bothmer, A.; Callen, E.; Reina-San-Martin, B.; Dorsett, Y.; Difilippantonio, S.; Bolland, D.J.; Chen, H.T.; Corcoran, A.E.; Nussenzweig, A.; et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell 2008, 135, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, E.; Begue-Pastor, N.; Chaganti, S.; Croom-Carter, D.; Shannon-Lowe, C.; Kube, D.; Feederle, R.; Delecluse, H.J.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus infection of naïve B cells in vitro frequently selects clones with mutated immunoglobulin genotypes: Implications for virus biology. PLoS Pathog. 2012, 8, e1002697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, A.; Folk, W.P.; Sakamuro, D. The Dual Roles of MYC in Genomic Instability and Cancer Chemoresistance. Genes 2017, 8, 158. [Google Scholar] [CrossRef] [Green Version]
- Allday, M.J. How does Epstein-Barr virus (EBV) complement the activation of Myc in the pathogenesis of Burkitt’s lymphoma? Semin. Cancer Biol. 2009, 19, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Jiang, C.; Zhang, Y.; Govande, A.; Trudeau, S.J.; Chen, F.; Fry, C.J.; Puri, R.; Wolinsky, E.; Schineller, M.; et al. MYC Controls the Epstein-Barr Virus Lytic Switch. Mol. Cell 2020, 78, 653–669.e658. [Google Scholar] [CrossRef]
- Neri, A.; Barriga, F.; Inghirami, G.; Knowles, D.M.; Neequaye, J.; Magrath, I.T.; Dalla-Favera, R. Epstein-Barr virus infection precedes clonal expansion in Burkitt’s and acquired immunodeficiency syndrome-associated lymphoma. Blood 1991, 77, 1092–1095. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, G.; Komano, J.; Sugden, B. Epstein-Barr virus provides a survival factor to Burkitt’s lymphomas. Proc. Natl. Acad. Sci. USA 2003, 100, 14269–14274. [Google Scholar] [CrossRef] [Green Version]
- Holowaty, M.N.; Frappier, L. HAUSP/USP7 as an Epstein-Barr virus target. Biochem. Soc. Trans. 2004, 32, 731–732. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.B.; Bell, J.L.; Levine, A.J. Expression of Epstein-Barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. EMBO J. 1996, 15, 3117–3126. [Google Scholar] [CrossRef]
- Soldan, S.S.; Anderson, E.M.; Frase, D.M.; Zhang, Y.; Caruso, L.B.; Wang, Y.; Deakyne, J.S.; Gewurz, B.E.; Tempera, I.; Lieberman, P.M.; et al. EBNA1 inhibitors have potent and selective antitumor activity in xenograft models of Epstein-Barr virus-associated gastric cancer. Gastric Cancer 2021, 24, 1076–1088. [Google Scholar] [CrossRef]
- Komano, J.; Maruo, S.; Kurozumi, K.; Oda, T.; Takada, K. Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt’s lymphoma cell line Akata. J. Virol. 1999, 73, 9827–9831. [Google Scholar] [CrossRef] [Green Version]
- Vereide, D.T.; Seto, E.; Chiu, Y.F.; Hayes, M.; Tagawa, T.; Grundhoff, A.; Hammerschmidt, W.; Sugden, B. Epstein-Barr virus maintains lymphomas via its miRNAs. Oncogene 2014, 33, 1258–1264. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Andreansky, S.; Diaz, G.; Turner, S.J.; Wodarz, D.; Doherty, P.C. Quantitative analysis of long-term virus-specific CD8+-T-cell memory in mice challenged with unrelated pathogens. J. Virol. 2003, 77, 7756–7763. [Google Scholar] [CrossRef] [Green Version]
- Moormann, A.M.; Chelimo, K.; Sumba, O.P.; Lutzke, M.L.; Ploutz-Snyder, R.; Newton, D.; Kazura, J.; Rochford, R. Exposure to holoendemic malaria results in elevated Epstein-Barr virus loads in children. J. Infect. Dis. 2005, 191, 1233–1238. [Google Scholar] [CrossRef] [Green Version]
- Moormann, A.M.; Bailey, J.A. Malaria—How this parasitic infection aids and abets EBV-associated Burkitt lymphomagenesis. Curr. Opin. Virol. 2016, 20, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Wilmore, J.R.; Maue, A.C.; Rochford, R. Plasmodium chabaudi infection induces AID expression in transitional and marginal zone B cells. Immun. Inflamm. Dis. 2016, 4, 497–505. [Google Scholar] [CrossRef]
- Torgbor, C.; Awuah, P.; Deitsch, K.; Kalantari, P.; Duca, K.A.; Thorley-Lawson, D.A. A multifactorial role for P. falciparum malaria in endemic Burkitt’s lymphoma pathogenesis. PLoS Pathog. 2014, 10, e1004170. [Google Scholar] [CrossRef] [Green Version]
- Robbiani, D.F.; Deroubaix, S.; Feldhahn, N.; Oliveira, T.Y.; Callen, E.; Wang, Q.; Jankovic, M.; Silva, I.T.; Rommel, P.C.; Bosque, D.; et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 2015, 162, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Mundo, L.; Ambrosio, M.R.; Picciolini, M.; Lo Bello, G.; Gazaneo, S.; Del Porro, L.; Lazzi, S.; Navari, M.; Onyango, N.; Granai, M.; et al. Unveiling Another Missing Piece in EBV-Driven Lymphomagenesis: EBV-Encoded MicroRNAs Expression in EBER-Negative Burkitt Lymphoma Cases. Front. Microbiol. 2017, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, R.L.; Chakravorty, A.; Sugden, B. Burkitt Lymphomas Evolve to Escape Dependencies on Epstein-Barr Virus. Front. Cell Infect. Microbiol. 2020, 10, 606412. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [Green Version]
- Giulino-Roth, L.; Wang, K.; MacDonald, T.Y.; Mathew, S.; Tam, Y.; Cronin, M.T.; Palmer, G.; Lucena-Silva, N.; Pedrosa, F.; Pedrosa, M.; et al. Targeted genomic sequencing of pediatric Burkitt lymphoma identifies recurrent alterations in antiapoptotic and chromatin-remodeling genes. Blood 2012, 120, 5181–5184. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Salles, G. Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2021, 384, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Shibusawa, M.; Kidoguchi, K.; Tanimoto, T. Epstein-Barr Virus-Positive Diffuse Large B Cell Lymphoma. In Lymphoma; Gallamini, A., Juweid, M., Eds.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar] [CrossRef]
- Oyama, T.; Yamamoto, K.; Asano, N.; Oshiro, A.; Suzuki, R.; Kagami, Y.; Morishima, Y.; Takeuchi, K.; Izumo, T.; Mori, S.; et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: A study of 96 patients. Clin. Cancer Res. 2007, 13, 5124–5132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ok, C.Y.; Papathomas, T.G.; Medeiros, L.J.; Young, K.H. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood 2013, 122, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Montes-Moreno, S.; Odqvist, L.; Diaz-Perez, J.A.; Lopez, A.B.; de Villambrosía, S.G.; Mazorra, F.; Castillo, M.E.; Lopez, M.; Pajares, R.; García, J.F.; et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod. Pathol. 2012, 25, 968–982. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Narbaitz, M.; Metrebian, F.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Epstein-Barr virus-positive diffuse large B-cell lymphoma association is not only restricted to elderly patients. Int. J. Cancer 2014, 135, 2816–2824. [Google Scholar] [CrossRef]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.; Xu, M.L.; Yu, H.; Fletcher, C.D.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef]
- Gebauer, N.; Künstner, A.; Ketzer, J.; Witte, H.M.; Rausch, T.; Benes, V.; Zimmermann, J.; Gebauer, J.; Merz, H.; Bernard, V.; et al. Genomic insights into the pathogenesis of Epstein-Barr virus-associated diffuse large B-cell lymphoma by whole-genome and targeted amplicon sequencing. Blood Cancer J. 2021, 11, 102. [Google Scholar] [CrossRef]
- Chabay, P. Advances in the Pathogenesis of EBV-Associated Diffuse Large B Cell Lymphoma. Cancers 2021, 13, 2717. [Google Scholar] [CrossRef]
- Kato, H.; Karube, K.; Yamamoto, K.; Takizawa, J.; Tsuzuki, S.; Yatabe, Y.; Kanda, T.; Katayama, M.; Ozawa, Y.; Ishitsuka, K.; et al. Gene expression profiling of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly reveals alterations of characteristic oncogenetic pathways. Cancer Sci. 2014, 105, 537–544. [Google Scholar] [CrossRef]
- Kataoka, K.; Miyoshi, H.; Sakata, S.; Dobashi, A.; Couronné, L.; Kogure, Y.; Sato, Y.; Nishida, K.; Gion, Y.; Shiraishi, Y.; et al. Frequent structural variations involving programmed death ligands in Epstein-Barr virus-associated lymphomas. Leukemia 2019, 33, 1687–1699. [Google Scholar] [CrossRef] [Green Version]
- Reusch, J.A.; Nawandar, D.M.; Wright, K.L.; Kenney, S.C.; Mertz, J.E. Cellular differentiation regulator BLIMP1 induces Epstein-Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J. Virol. 2015, 89, 1731–1743. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Küppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc. Natl. Acad. Sci. USA 1994, 91, 10962–10966. [Google Scholar] [CrossRef] [Green Version]
- Küppers, R.; Hansmann, M.L. The Hodgkin and Reed/Sternberg cell. Int. J. Biochem. Cell Biol. 2005, 37, 511–517. [Google Scholar] [CrossRef]
- Fornatora, M.; Reich, R.F.; Freedman, P. Extranodal Hodgkin’s lymphoma of the oral soft tissue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004, 98, 207–208; [Google Scholar] [CrossRef]
- Shanbhag, S.; Ambinder, R.F. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J. Clin. 2018, 68, 116–132. [Google Scholar] [CrossRef]
- Glaser, S.L.; Lin, R.J.; Stewart, S.L.; Ambinder, R.F.; Jarrett, R.F.; Brousset, P.; Pallesen, G.; Gulley, M.L.; Khan, G.; O’Grady, J.; et al. Epstein-Barr virus-associated Hodgkin’s disease: Epidemiologic characteristics in international data. Int. J. Cancer 1997, 70, 375–382. [Google Scholar] [CrossRef]
- Hjalgrim, H.; Askling, J.; Rostgaard, K.; Hamilton-Dutoit, S.; Frisch, M.; Zhang, J.S.; Madsen, M.; Rosdahl, N.; Konradsen, H.B.; Storm, H.H.; et al. Characteristics of Hodgkin’s lymphoma after infectious mononucleosis. N. Engl. J. Med. 2003, 349, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Hanson, D.L.; Sullivan, P.S.; Novak, R.M.; Moorman, A.C.; Tong, T.C.; Holmberg, S.D.; Brooks, J.T. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992–2003. Ann. Intern. Med. 2008, 148, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanzler, H.; Küppers, R.; Hansmann, M.L.; Rajewsky, K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Bräuninger, A.; Schmitz, R.; Bechtel, D.; Renné, C.; Hansmann, M.L.; Küppers, R. Molecular biology of Hodgkin’s and Reed/Sternberg cells in Hodgkin’s lymphoma. Int. J. Cancer 2006, 118, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.A.; Küppers, R. Molecular biology of Hodgkin lymphoma. Leukemia 2021, 35, 968–981. [Google Scholar] [CrossRef] [PubMed]
- Floettmann, J.E.; Rowe, M. Epstein-Barr virus latent membrane protein-1 (LMP1) C-terminus activation region 2 (CTAR2) maps to the far C-terminus and requires oligomerisation for NF-kappaB activation. Oncogene 1997, 15, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Vockerodt, M.; Morgan, S.L.; Kuo, M.; Wei, W.; Chukwuma, M.B.; Arrand, J.R.; Kube, D.; Gordon, J.; Young, L.S.; Woodman, C.B.; et al. The Epstein-Barr virus oncoprotein, latent membrane protein-1, reprograms germinal centre B cells towards a Hodgkin’s Reed-Sternberg-like phenotype. J. Pathol. 2008, 216, 83–92. [Google Scholar] [CrossRef]
- Vrzalikova, K.; Vockerodt, M.; Leonard, S.; Bell, A.; Wei, W.; Schrader, A.; Wright, K.L.; Kube, D.; Rowe, M.; Woodman, C.B.; et al. Down-regulation of BLIMP1α by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: Implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 2011, 117, 5907–5917. [Google Scholar] [CrossRef]
- Mancao, C.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, H.; Yamakawa, N.; Imadome, K.I.; Yahata, T.; Kotaki, R.; Ogata, J.; Kakizaki, M.; Fujita, K.; Lu, J.; Yokoyama, K.; et al. Role of exosomes as a proinflammatory mediator in the development of EBV-associated lymphoma. Blood 2018, 131, 2552–2567. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Celegato, M.; Casagrande, N. Microenvironmental interactions in classical Hodgkin lymphoma and their role in promoting tumor growth, immune escape and drug resistance. Cancer Lett. 2016, 380, 243–252. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Gruss, H.J.; Pinto, A. CD40 ligand is constitutively expressed in a subset of T cell lymphomas and on the microenvironmental reactive T cells of follicular lymphomas and Hodgkin’s disease. Am. J. Pathol. 1995, 147, 912–922. [Google Scholar]
- Assis, M.C.; Campos, A.H.; Oliveira, J.S.; Soares, F.A.; Silva, J.M.; Silva, P.B.; Penna, A.D.; Souza, E.M.; Baiocchi, O.C. Increased expression of CD4+CD25 +FOXP3+ regulatory T cells correlates with Epstein-Barr virus and has no impact on survival in patients with classical Hodgkin lymphoma in Brazil. Med. Oncol. 2012, 29, 3614–3619. [Google Scholar] [CrossRef]
- Wienand, K.; Chapuy, B.; Stewart, C.; Dunford, A.J.; Wu, D.; Kim, J.; Kamburov, A.; Wood, T.R.; Cader, F.Z.; Ducar, M.D.; et al. Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv 2019, 3, 4065–4080. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.S. Epstein-Barr virus: A master epigenetic manipulator. Curr. Opin. Virol. 2017, 26, 74–80. [Google Scholar] [CrossRef]
- Johnson, N.W.; Jayasekara, P.; Amarasinghe, A.A. Squamous cell carcinoma and precursor lesions of the oral cavity: Epidemiology and aetiology. Periodontology 2011, 57, 19–37. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Butt, F.M.; Chindia, M.L.; Rana, F. Oral squamous cell carcinoma in human immunodeficiency virus positive patients: Clinicopathological audit. J. Laryngol. Otol. 2012, 126, 276–278. [Google Scholar] [CrossRef] [Green Version]
- SEER Cancer Stat Facts: Oral Cavity and Pharynx Cancer. Available online: https://seer.cancer.gov/statfacts/html/oralcav.html (accessed on 25 October 2022).
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Guidry, J.T.; Birdwell, C.E.; Scott, R.S. Epstein-Barr virus in the pathogenesis of oral cancers. Oral Dis. 2018, 24, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Rahman, R.; Gopinath, D.; Buajeeb, W.; Poomsawat, S.; Johnson, N.W. Potential Role of Epstein-Barr Virus in Oral Potentially Malignant Disorders and Oral Squamous Cell Carcinoma: A Scoping Review. Viruses 2022, 14, 801. [Google Scholar] [CrossRef]
- Bavle, R.M.; Venugopal, R.; Konda, P.; Muniswamappa, S.; Makarla, S. Molecular Classification of Oral Squamous Cell Carcinoma. J. Clin. Diagn. Res. 2016, 10, Ze18–Ze21. [Google Scholar] [CrossRef]
- Kikuchi, K.; Noguchi, Y.; de Rivera, M.W.; Hoshino, M.; Sakashita, H.; Yamada, T.; Inoue, H.; Miyazaki, Y.; Nozaki, T.; González-López, B.S.; et al. Detection of Epstein-Barr virus genome and latent infection gene expression in normal epithelia, epithelial dysplasia, and squamous cell carcinoma of the oral cavity. Tumour Biol. 2016, 37, 3389–3404. [Google Scholar] [CrossRef]
- She, Y.; Nong, X.; Zhang, M.; Wang, M. Epstein-Barr virus infection and oral squamous cell carcinoma risk: A meta-analysis. PLoS ONE 2017, 12, e0186860. [Google Scholar] [CrossRef] [Green Version]
- Pathmanathan, R.; Prasad, U.; Sadler, R.; Flynn, K.; Raab-Traub, N. Clonal proliferations of cells infected with Epstein-Barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N. Engl. J. Med. 1995, 333, 693–698. [Google Scholar] [CrossRef]
- Rahman, R.; Poomsawat, S.; Juengsomjit, R.; Buajeeb, W. Overexpression of Epstein-Barr virus-encoded latent membrane protein-1 (LMP-1) in oral squamous cell carcinoma. BMC Oral Health 2019, 19, 142. [Google Scholar] [CrossRef]
- Slots, J.; Contreras, A. Herpesviruses: A unifying causative factor in periodontitis? Oral Microbiol. Immunol. 2000, 15, 277–280. [Google Scholar] [CrossRef]
- Imai, K.; Inoue, H.; Tamura, M.; Cueno, M.E.; Takeichi, O.; Kusama, K.; Saito, I.; Ochiai, K. The periodontal pathogen Porphyromonas gingivalis induces the Epstein-Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie 2012, 94, 839–846. [Google Scholar] [CrossRef]
- Birdwell, C.E.; Queen, K.J.; Kilgore, P.C.; Rollyson, P.; Trutschl, M.; Cvek, U.; Scott, R.S. Genome-wide DNA methylation as an epigenetic consequence of Epstein-Barr virus infection of immortalized keratinocytes. J. Virol. 2014, 88, 11442–11458. [Google Scholar] [CrossRef] [Green Version]
- Scholle, F.; Bendt, K.M.; Raab-Traub, N. Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J. Virol. 2000, 74, 10681–10689. [Google Scholar] [CrossRef] [Green Version]
- Romero-Masters, J.C.; Ohashi, M.; Djavadian, R.; Eichelberg, M.R.; Hayes, M.; Zumwalde, N.A.; Bristol, J.A.; Nelson, S.E.; Ma, S.; Ranheim, E.A.; et al. An EBNA3A-Mutated Epstein-Barr Virus Retains the Capacity for Lymphomagenesis in a Cord Blood-Humanized Mouse Model. J. Virol. 2020, 94, e02168-19. [Google Scholar] [CrossRef]
- Queen, K.J.; Shi, M.; Zhang, F.; Cvek, U.; Scott, R.S. Epstein-Barr virus-induced epigenetic alterations following transient infection. Int. J. Cancer 2013, 132, 2076–2086. [Google Scholar] [CrossRef] [Green Version]
- Shair, K.H.; Schnegg, C.I.; Raab-Traub, N. EBV latent membrane protein 1 effects on plakoglobin, cell growth, and migration. Cancer Res. 2008, 68, 6997–7005. [Google Scholar] [CrossRef] [Green Version]
- Dawson, C.W.; Laverick, L.; Morris, M.A.; Tramoutanis, G.; Young, L.S. Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J. Virol. 2008, 82, 3654–3664. [Google Scholar] [CrossRef] [Green Version]
- Mainou, B.A.; Raab-Traub, N. LMP1 strain variants: Biological and molecular properties. J. Virol. 2006, 80, 6458–6468. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jia, L.; Liu, T.; Yip, Y.L.; Tang, W.C.; Lin, W.; Deng, W.; Lo, K.W.; You, C.; Lung, M.L.; et al. mTORC2-mediated PDHE1α nuclear translocation links EBV-LMP1 reprogrammed glucose metabolism to cancer metastasis in nasopharyngeal carcinoma. Oncogene 2019, 38, 4669–4684. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramqvist, T.; Dalianis, T. Oropharyngeal cancer epidemic and human papillomavirus. Emerg. Infect. Dis. 2010, 16, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Scott-Wittenborn, N.; Fakhry, C. Epidemiology of HPV Related Malignancies. Semin. Radiat. Oncol. 2021, 31, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Jones, O.S.; Breeze, C.E.; Gilson, R. Gender-neutral HPV vaccination in the UK, rising male oropharyngeal cancer rates, and lack of HPV awareness. Lancet Infect. Dis. 2019, 19, 131–132. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.; Liu, J.; Masterson, L.; Fenton, T.R. HPV-associated oropharyngeal cancer: Epidemiology, molecular biology and clinical management. Nat. Rev. Clin. Oncol. 2022, 19, 306–327. [Google Scholar] [CrossRef]
- Faraji, F.; Rettig, E.M.; Tsai, H.L.; El Asmar, M.; Fung, N.; Eisele, D.W.; Fakhry, C. The prevalence of human papillomavirus in oropharyngeal cancer is increasing regardless of sex or race, and the influence of sex and race on survival is modified by human papillomavirus tumor status. Cancer 2019, 125, 761–769. [Google Scholar] [CrossRef]
- Taberna, M.; Mena, M.; Pavón, M.A.; Alemany, L.; Gillison, M.L.; Mesía, R. Human papillomavirus-related oropharyngeal cancer. Ann. Oncol. 2017, 28, 2386–2398. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Bai, Y.P.; Mao, M.L.; Zhang, H.; Zhao, X.L.; Yang, D.M.; Wan, H.F.; Liu, H.G. Clinicopathological characteristics of HPV(+) oropharyngeal squamous cell carcinoma. Chin. J. Pathol. 2019, 48, 127–131. [Google Scholar] [CrossRef]
- Upile, N.S.; Shaw, R.J.; Jones, T.M.; Goodyear, P.; Liloglou, T.; Risk, J.M.; Boyd, M.T.; Sheard, J.; Sloan, P.; Robinson, M.; et al. Squamous cell carcinoma of the head and neck outside the oropharynx is rarely human papillomavirus related. Laryngoscope 2014, 124, 2739–2744. [Google Scholar] [CrossRef]
- Yete, S.; D’Souza, W.; Saranath, D. High-Risk Human Papillomavirus in Oral Cancer: Clinical Implications. Oncology 2018, 94, 133–141. [Google Scholar] [CrossRef]
- Jalouli, J.; Jalouli, M.M.; Sapkota, D.; Ibrahim, S.O.; Larsson, P.A.; Sand, L. Human papilloma virus, herpes simplex virus and epstein barr virus in oral squamous cell carcinoma from eight different countries. Anticancer Res. 2012, 32, 571–580. [Google Scholar]
- Polz-Gruszka, D.; Morshed, K.; Jarzyński, A.; Polz-Dacewicz, M. Prevalence of Polyoma BK Virus (BKPyV), Epstein-Barr Virus (EBV) and Human Papilloma Virus (HPV) in Oropharyngeal Cancer. Pol. J. Microbiol. 2015, 64, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Drop, B.; Strycharz-Dudziak, M.; Kliszczewska, E.; Polz-Dacewicz, M. Coinfection with Epstein-Barr Virus (EBV), Human Papilloma Virus (HPV) and Polyoma BK Virus (BKPyV) in Laryngeal, Oropharyngeal and Oral Cavity Cancer. Int. J. Mol. Sci. 2017, 18, 2752. [Google Scholar] [CrossRef] [Green Version]
- Broccolo, F.; Ciccarese, G.; Rossi, A.; Anselmi, L.; Drago, F.; Toniolo, A. Human papillomavirus (HPV) and Epstein-Barr virus (EBV) in keratinizing versus non- keratinizing squamous cell carcinoma of the oropharynx. Infect. Agents Cancer 2018, 13, 32. [Google Scholar] [CrossRef]
- Jiang, R.; Ekshyyan, O.; Moore-Medlin, T.; Rong, X.; Nathan, S.; Gu, X.; Abreo, F.; Rosenthal, E.L.; Shi, M.; Guidry, J.T.; et al. Association between human papilloma virus/Epstein-Barr virus coinfection and oral carcinogenesis. J. Oral Pathol. Med. 2015, 44, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Prayitno, A. Cervical cancer with human papilloma virus and Epstein Barr virus positive. J. Carcinog. 2006, 5, 13. [Google Scholar] [CrossRef]
- Szostek, S.; Zawilinska, B.; Kopec, J.; Kosz-Vnenchak, M. Herpesviruses as possible cofactors in HPV-16-related oncogenesis. Acta Biochim. Pol. 2009, 56, 337–342. [Google Scholar] [CrossRef] [Green Version]
- De Lima, M.A.P.; Neto, P.J.N.; Lima, L.P.M.; Gonçalves Júnior, J.; Teixeira Junior, A.G.; Teodoro, I.P.P.; Facundo, H.T.; da Silva, C.G.L.; Lima, M.V.A. Association between Epstein-Barr virus (EBV) and cervical carcinoma: A meta-analysis. Gynecol. Oncol. 2018, 148, 317–328. [Google Scholar] [CrossRef]
- Kahla, S.; Oueslati, S.; Achour, M.; Kochbati, L.; Chanoufi, M.B.; Maalej, M.; Oueslati, R. Correlation between ebv co-infection and HPV16 genome integrity in Tunisian cervical cancer patients. Braz. J. Microbiol. 2012, 43, 744–753. [Google Scholar] [CrossRef] [Green Version]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raab-Traub, N.; Flynn, K. The structure of the termini of the Epstein-Barr virus as a marker of clonal cellular proliferation. Cell 1986, 47, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Scott, R.S.; Su, T.; Sixbey, J.W. Length of Epstein-Barr virus termini as a determinant of epithelial cell clonal emergence. J. Virol. 2003, 77, 8555–8561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repic, A.M.; Shi, M.; Scott, R.S.; Sixbey, J.W. Augmented latent membrane protein 1 expression from Epstein-Barr virus episomes with minimal terminal repeats. J. Virol. 2010, 84, 2236–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidry, J.T.; Myers, J.E.; Bienkowska-Haba, M.; Songock, W.K.; Ma, X.; Shi, M.; Nathan, C.O.; Bodily, J.M.; Sapp, M.J.; Scott, R.S. Inhibition of Epstein-Barr Virus Replication in Human Papillomavirus-Immortalized Keratinocytes. J. Virol. 2019, 93, e01216-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar]
- Makielski, K.R.; Lee, D.; Lorenz, L.D.; Nawandar, D.M.; Chiu, Y.F.; Kenney, S.C.; Lambert, P.F. Human papillomavirus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes. Virology 2016, 495, 52–62. [Google Scholar] [CrossRef]
- Gillison, M.L.; Akagi, K.; Xiao, W.; Jiang, B.; Pickard, R.K.L.; Li, J.; Swanson, B.J.; Agrawal, A.D.; Zucker, M.; Stache-Crain, B.; et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2019, 29, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hafkamp, H.C.; Speel, E.J.; Haesevoets, A.; Bot, F.J.; Dinjens, W.N.; Ramaekers, F.C.; Hopman, A.H.; Manni, J.J. A subset of head and neck squamous cell carcinomas exhibits integration of HPV 16/18 DNA and overexpression of p16INK4A and p53 in the absence of mutations in p53 exons 5–8. Int. J. Cancer 2003, 107, 394–400. [Google Scholar] [CrossRef]
- Li, W.; Thompson, C.H.; O’Brien, C.J.; McNeil, E.B.; Scolyer, R.A.; Cossart, Y.E.; Veness, M.J.; Walker, D.M.; Morgan, G.J.; Rose, B.R. Human papillomavirus positivity predicts favourable outcome for squamous carcinoma of the tonsil. Int. J. Cancer 2003, 106, 553–558. [Google Scholar] [CrossRef]
- Fischer, C.A.; Zlobec, I.; Green, E.; Probst, S.; Storck, C.; Lugli, A.; Tornillo, L.; Wolfensberger, M.; Terracciano, L.M. Is the improved prognosis of p16 positive oropharyngeal squamous cell carcinoma dependent of the treatment modality? Int. J. Cancer 2010, 126, 1256–1262. [Google Scholar] [CrossRef]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Zhu, B.; Xiao, Y.; Yeager, M.; Clifford, G.; Wentzensen, N.; Cullen, M.; Boland, J.F.; Bass, S.; Steinberg, M.K.; Raine-Bennett, T.; et al. Mutations in the HPV16 genome induced by APOBEC3 are associated with viral clearance. Nat. Commun. 2020, 11, 886. [Google Scholar] [CrossRef] [Green Version]
- Schiødt, M.; Greenspan, D.; Daniels, T.E.; Greenspan, J.S. Clinical and histologic spectrum of oral hairy leukoplakia. Oral Surg. Oral Med. Oral Pathol. 1987, 64, 716–720. [Google Scholar] [CrossRef]
- Greenspan, J.S.; Greenspan, D. Oral hairy leukoplakia: Diagnosis and management. Oral Surg. Oral Med. Oral Pathol. 1989, 67, 396–403. [Google Scholar] [CrossRef]
- Greenspan, D.; Greenspan, J.S.; Conant, M.; Petersen, V.; Silverman, S., Jr.; de Souza, Y. Oral “hairy” leucoplakia in male homosexuals: Evidence of association with both papillomavirus and a herpes-group virus. Lancet 1984, 2, 831–834. [Google Scholar] [CrossRef]
- Rathee, M.; Jain, P. Hairy Leukoplakia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://pubmed.ncbi.nlm.nih.gov/32491354/ (accessed on 16 June 2022).
- Piperi, E.; Omlie, J.; Koutlas, I.G.; Pambuccian, S. Oral hairy leukoplakia in HIV-negative patients: Report of 10 cases. Int. J. Surg. Pathol. 2010, 18, 177–183. [Google Scholar] [CrossRef]
- Greenspan, J.S.; De Souza, Y.G.; Regezi, J.A.; Daniels, T.E.; Greenspan, D.; MacPhail, L.A.; Hilton, J.F. Comparison of cytopathic changes in oral hairy leukoplakia with in situ hybridization for EBV DNA. Oral Dis. 1998, 4, 95–99. [Google Scholar] [CrossRef]
- Webster-Cyriaque, J.; Middeldorp, J.; Raab-Traub, N. Hairy leukoplakia: An unusual combination of transforming and permissive Epstein-Barr virus infections. J. Virol. 2000, 74, 7610–7618. [Google Scholar] [CrossRef] [Green Version]
- Husak, R.; Garbe, C.; Orfanos, C.E. Oral hairy leukoplakia in 71 HIV-seropositive patients: Clinical symptoms, relation to immunologic status, and prognostic significance. J. Am. Acad. Dermatol. 1996, 35, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Walling, D.M. Oral hairy leukoplakia: An Epstein-Barr virus-associated disease of patients with HIV. Res. Initiat. Treat. Action 2000, 6, 10–15. [Google Scholar] [PubMed]
- Tugizov, S.; Herrera, R.; Veluppillai, P.; Greenspan, J.; Greenspan, D.; Palefsky, J.M. Epstein-Barr virus (EBV)-infected monocytes facilitate dissemination of EBV within the oral mucosal epithelium. J. Virol. 2007, 81, 5484–5496. [Google Scholar] [CrossRef] [PubMed]
- Resnick, L.; Herbst, J.S.; Ablashi, D.V.; Atherton, S.; Frank, B.; Rosen, L.; Horwitz, S.N. Regression of oral hairy leukoplakia after orally administered acyclovir therapy. JAMA 1988, 259, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Felix, D.H.; Wray, D.; Southam, J.C.; Cubie, H.A.; Crawford, D.H. Epstein-Barr virus gene expression and epithelial cell differentiation in oral hairy leukoplakia. Am. J. Pathol. 1991, 139, 1369–1380. [Google Scholar]
- Walling, D.M.; Flaitz, C.M.; Hosein, F.G.; Montes-Walters, M.; Nichols, C.M. Effect of Epstein-Barr virus replication on Langerhans cells in pathogenesis of oral hairy leukoplakia. J. Infect. Dis. 2004, 189, 1656–1663. [Google Scholar] [CrossRef] [Green Version]
- Dawson, C.W.; Eliopoulos, A.G.; Dawson, J.; Young, L.S. BHRF1, a viral homologue of the Bcl-2 oncogene, disturbs epithelial cell differentiation. Oncogene 1995, 10, 69–77. [Google Scholar]
- Walling, D.M.; Edmiston, S.N.; Sixbey, J.W.; Abdel-Hamid, M.; Resnick, L.; Raab-Traub, N. Coinfection with multiple strains of the Epstein-Barr virus in human immunodeficiency virus-associated hairy leukoplakia. Proc. Natl. Acad. Sci. USA 1992, 89, 6560–6564. [Google Scholar] [CrossRef] [Green Version]
- Walling, D.M.; Shebib, N.; Weaver, S.C.; Nichols, C.M.; Flaitz, C.M.; Webster-Cyriaque, J. The molecular epidemiology and evolution of Epstein-Barr virus: Sequence variation and genetic recombination in the latent membrane protein-1 gene. J. Infect. Dis. 1999, 179, 763–774. [Google Scholar] [CrossRef]
- Webster-Cyriaque, J.; Raab-Traub, N. Transcription of Epstein-Barr virus latent cycle genes in oral hairy leukoplakia. Virology 1998, 248, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Damania, B.; Kenney, S.C.; Raab-Traub, N. Epstein-Barr virus: Biology and clinical disease. Cell 2022, 185, 3652–3670. [Google Scholar] [CrossRef]
- Hamilton-Dutoit, S.J.; Therkildsen, M.H.; Neilsen, N.H.; Jensen, H.; Hansen, J.P.; Pallesen, G. Undifferentiated carcinoma of the salivary gland in Greenlandic Eskimos: Demonstration of Epstein-Barr virus DNA by in situ nucleic acid hybridization. Hum. Pathol. 1991, 22, 811–815. [Google Scholar] [CrossRef]
- Iezzoni, J.C.; Gaffey, M.J.; Weiss, L.M. The role of Epstein-Barr virus in lymphoepithelioma-like carcinomas. Am. J. Clin. Pathol. 1995, 103, 308–315. [Google Scholar] [CrossRef]
- Leung, S.Y.; Chung, L.P.; Yuen, S.T.; Ho, C.M.; Wong, M.P.; Chan, S.Y. Lymphoepithelial carcinoma of the salivary gland: In situ detection of Epstein-Barr virus. J. Clin. Pathol. 1995, 48, 1022–1027. [Google Scholar] [CrossRef]
- Kuo, T.; Hsueh, C. Lymphoepithelioma-like salivary gland carcinoma in Taiwan: A clinicopathological study of nine cases demonstrating a strong association with Epstein-Barr virus. Histopathology 1997, 31, 75–82. [Google Scholar] [CrossRef]
- Jen, K.Y.; Cheng, J.; Li, J.; Wu, L.; Li, Y.; Yu, S.; Lin, H.; Chen, Z.; Gurtsevitch, V.; Saku, T. Mutational events in LMP1 gene of Epstein-Barr virus in salivary gland lymphoepithelial carcinomas. Int. J. Cancer 2003, 105, 654–660. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cancer | Cellular Origin | % EBV Associated | EBV Expression Pattern | Latent Gene Products Detected |
|---|---|---|---|---|
| Burkitt lymphoma Endemic Sporadic HIV-related | Germinal center centroblast | ~100% 10–80% 30–40% | Latency I | EBERs, EBNA1, BARTs |
| Hodgkin lymphoma-Classic variant Nodular sclerosis Mixed cellularity HIV-related Hodgkin lymphoma-NLPHL variant | Post-germinal center centroblast | 40–50% 10–40% 70–80% >90% rare | Latency II | EBERs, EBNA1, LMP1, LMP2, BARTs |
| DLBCL NOS HIV-related, centroblastic HIV-related, immunoblastic | Post-germinal center centroblast | 3–50% * 30% 90% | Latency II/III Latency I/II/III Latency I/II/III | Depends on latency program |
| Oral Squamous Cell Carcinoma | Epithelial Cells | 0–80% | EBERs, EBNA2, LMP1 | |
| HPV-positive oropharyngeal squamous cell carcinoma | Epithelial cells | 5–25% | EBERs | |
| Oral Hairy Leukoplakia | Epithelial Cells | 100% | Lytic | All viral genes |
| Salivary Gland Epithelioma | Epithelial Cells | EBERs, LMP1 |
| EBV Latent Gene Product | Function |
|---|---|
| EBNA1 | Required for viral genome latent replication and segregation Promotes resistance to apoptosis by degradation of p53 Increases ROS production and genomic instability |
| EBNA2 | Essential for B cell immortalization Regulates viral and host gene expression by interacting with host transcription factors and EBNALP Regulates chromatin looping and accessibility |
| EBNA3A/C | Recruits polycomb repressor complex 2 for epigenetic repression of cyclin dependent kinase inhibitors (CKIs) and apoptotic factors Induces AID expression (EBNA 3C) Promotes bypasses cell cycle checkpoints that increase proliferation and genomic instability |
| EBNA3B | Tumor suppressor activity |
| EBNALP | Transcriptional co-activator of EBNA2 |
| LMP1 | Mimics CD40 receptor signaling Activates NF-kB/MAPK/JAK-STAT/PI3K signaling Induces DNA methyltransferase activity Promotes proliferation and survival Induces AID expression Immune modulation |
| LMP2A/B | Mimics host B cell receptor (BCR) signaling Blocks tyrosine kinase signaling following antigen activation of BCR Inhibits viral reactivation Induces DNA methyltransferase activity Enhances cell migration Inhibits epithelial differentiation |
| EBER1 | Abundantly expressed viral RNA in EBV latency Confers resistance to apoptosis Retains cellular ribosomal factor L22 in the nucleoplasm Blocks interferon inducible protein kinase R (PKR)-mediated inhibition of protein synthesis |
| EBER2 | Binds and recruits Pax5 to EBV terminal repeats |
| BART microRNAs | Increases resistance to apoptosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, B.J.H.; Schaal, D.L.; Nkadi, E.H.; Scott, R.S. EBV Association with Lymphomas and Carcinomas in the Oral Compartment. Viruses 2022, 14, 2700. https://doi.org/10.3390/v14122700
Ward BJH, Schaal DL, Nkadi EH, Scott RS. EBV Association with Lymphomas and Carcinomas in the Oral Compartment. Viruses. 2022; 14(12):2700. https://doi.org/10.3390/v14122700
Chicago/Turabian StyleWard, B. J. H., Danielle L. Schaal, Ebubechukwu H. Nkadi, and Rona S. Scott. 2022. "EBV Association with Lymphomas and Carcinomas in the Oral Compartment" Viruses 14, no. 12: 2700. https://doi.org/10.3390/v14122700
APA StyleWard, B. J. H., Schaal, D. L., Nkadi, E. H., & Scott, R. S. (2022). EBV Association with Lymphomas and Carcinomas in the Oral Compartment. Viruses, 14(12), 2700. https://doi.org/10.3390/v14122700