
Identifying Inhibitors of −1 Programmed Ribosomal Frameshifting in a Broad Spectrum of Coronaviruses

 , , ,
, , ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Results
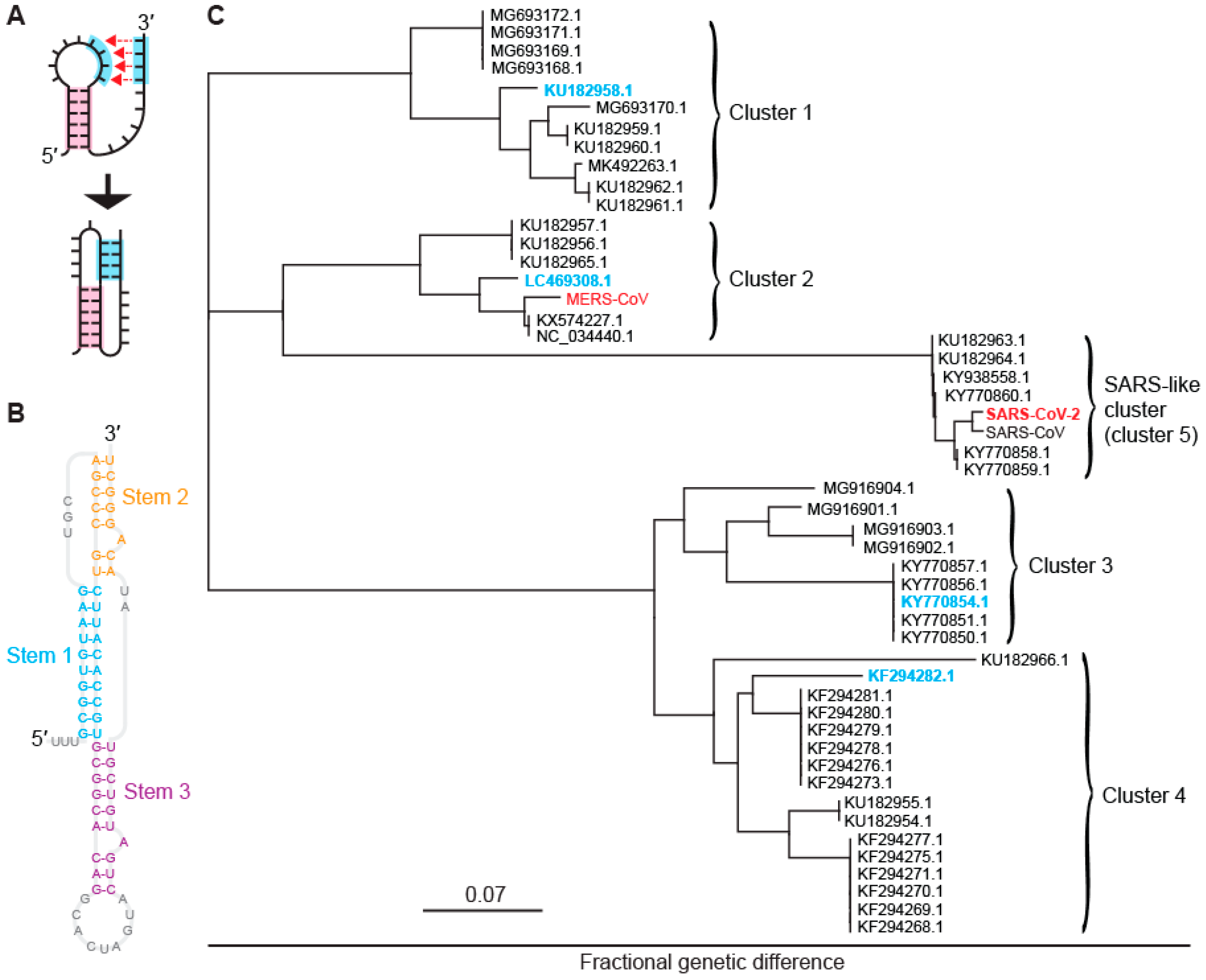
2.1. Identifying Five Distinct Clusters of −1 PRFsignals in Bat CoVs
2.2. Identifying Small-Molecule Modulators of SARS-CoV-2 −1 PRF
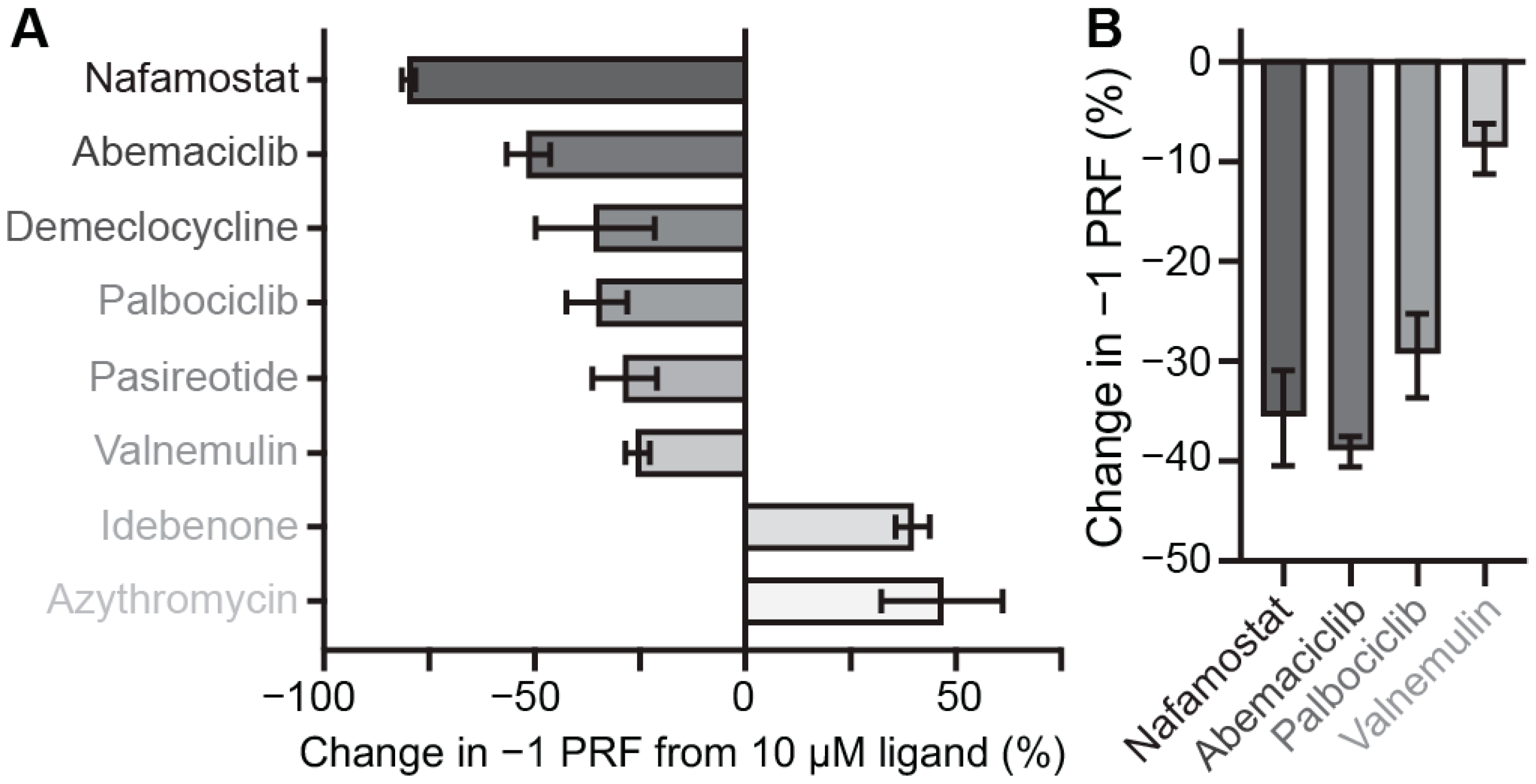
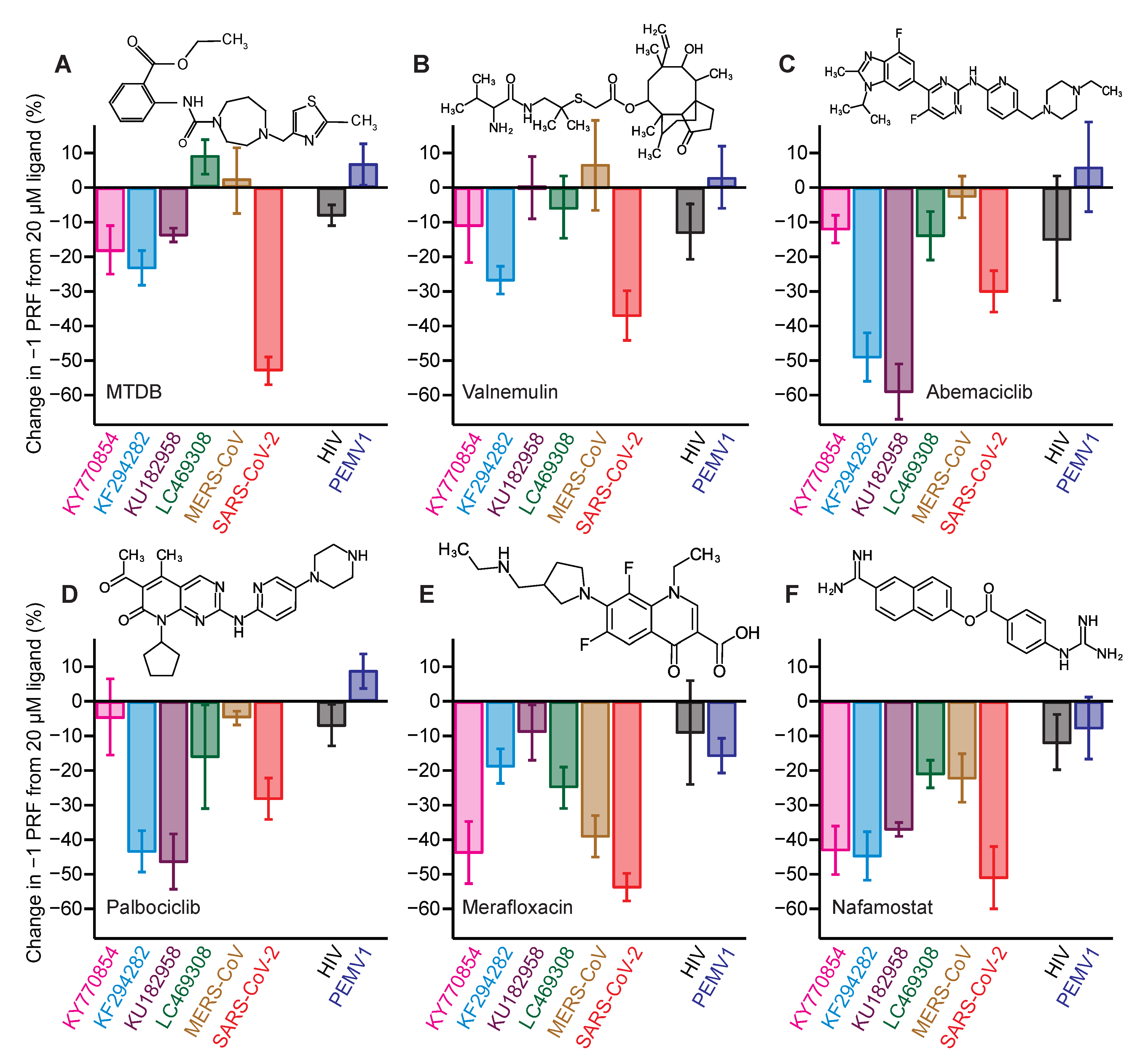
2.3. Identifying Pan-CoV −1 PRF Inhibitors
3. Discussion
4. Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weiss, S.R. Forty years with coronaviruses. J. Exp. Med. 2020, 217, e20200537. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.; Mossman, K. Bats and coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, T.J.; Cryan, P.; Cunningham, A.A.; Fooks, A.R.; Hayman, D.T.S.; Luis, A.D.; Peel, A.J.; Plowright, R.K.; Wood, J.L.N. Bat flight and zoonotic viruses. Emerging Infect. Dis. 2014, 20, 741–745. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.A.; Woodside, M.T.; Dinman, J.D. Programmed −1 Ribosomal Frameshifting in coronaviruses: A therapeutic target. Virology 2021, 554, 75–82. [Google Scholar] [CrossRef]
- Brierley, I.; Gilbert, R.J.; Pennell, S. Pseudoknot-dependent programmed −1 ribosomal frameshifting: Structures, mechanisms and models. In Recoding: Expansion of Decoding Rules Enriches Gene Expression; Springer: New York, NY, USA, 2010; pp. 149–174. [Google Scholar]
- Dinman, J.D. Mechanisms and implications of programmed translational frameshifting. Wiley Interdiscip. Rev. RNA. 2012, 3, 661–673. [Google Scholar] [CrossRef]
- Atkins, J.F.; Loughran, G.; Bhatt, P.R.; Firth, A.E.; Baranov, P.V. Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016, 44, 7007–7078. [Google Scholar] [CrossRef] [Green Version]
- Plant, E.P.; Rakauskaitė, R.; Taylor, D.R.; Dinman, J.D. Achieving a golden mean: Mechanisms by which coronaviruses ensure synthesis of the correct stoichiometric ratios of viral proteins. J. Virol. 2010, 84, 4330–4340. [Google Scholar] [CrossRef] [Green Version]
- Plant, E.P.; Sims, A.C.; Baric, R.S.; Dinman, J.D.; Taylor, D.R. Altering SARS coronavirus frameshift efficiency affects genomic and subgenomic RNAproduction. Viruses 2013, 5, 279–294. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, P.R.; Scaiola, A.; Loughran, G.; Leibundgut, M.; Kratzel, A.; Meurs, R.; Dreos, R.; O’Connor, K.M.; McMillan, A.; Bode, J.W.; et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science 2021, 372, 1306–1313. [Google Scholar] [CrossRef]
- Sun, Y.; Abriola, L.; Niederer, R.O.; Pedersen, S.F.; Alfajaro, M.M.; Monteiro, V.S.; Wilen, C.B.; Ho, Y.-C.; Gilbert, W.V.; Surovtseva, Y.V.; et al. Restriction of SARS-CoV-2 replication by targeting programmed −1 ribosomal frameshifting. Proc. Natl. Acad. Sci. USA 2021, 118, e2023051118. [Google Scholar] [CrossRef]
- Dinman, J.D.; Wickner, R.B. Ribosomal frameshifting efficiency and gag/gag-pol ratio are critical for yeast M1 double-stranded RNA virus propagation. J. Virol. 1992, 66, 3669–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehu-Xhilaga, M.; Crowe, S.M.; Mak, J. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 2001, 75, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
- Dulude, D.; Berchiche, Y.A.; Gendron, K.; Brakier-Gingras, L.; Heveker, N. Decreasing the frameshift efficiency translates into an equivalent reduction of the replication of the human immunodeficiency virus type 1. Virology 2006, 345, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Sztuba-Solinska, J.; Chavez-Calvillo, G.; Cline, S.E. Unveiling the druggable RNA targets and small molecule therapeutics. Biorg. Med. Chem. 2019, 27, 2149–2165. [Google Scholar] [CrossRef]
- Connelly, C.M.; Moon, M.H.; Schneekloth, J.S., Jr. The emerging role of RNA as a therapeutic target for small molecules. Cell Chem. Biol. 2016, 23, 1077–1090. [Google Scholar] [CrossRef]
- Plant, E.P.; Pérez-Alvarado, G.C.; Jacobs, J.L.; Mukhopadhyay, B.; Hennig, M.; Dinman, J.D. A three-stemmed mRNA pseudoknot in the SARS coronavirus frameshift signal. PLoS Biol. 2005, 3, e172. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.A.; Olson, A.N.; Neupane, K.; Munshi, S.; San Emeterio, J.; Pollack, L.; Woodside, M.T.; Dinman, J.D. Structural and functional conservation of the programmed −1 ribosomal frameshift signal of SARS coronavirus 2 (SARS-CoV-2). J. Biol. Chem. 2020, 295, 10741–10748. [Google Scholar] [CrossRef]
- Omar, S.I.; Zhao, M.; Sekar, R.V.; Moghadam, S.A.; Tuszynski, J.A.; Woodside, M.T. Modeling the structure of the frameshift-stimulatory pseudoknot in SARS-CoV-2 reveals multiple possible conformers. PLoS Comp. Biol. 2021, 17, e1008603. [Google Scholar] [CrossRef]
- Zhang, K.; Zheludev, I.N.; Hagey, R.J.; Haslecker, R.; Hou, Y.J.; Kretsch, R.; Pintilie, G.D.; Rangan, R.; Kladwang, W.; Li, S.; et al. Cryo-EM and antisense targeting of the 28-kDa frameshift stimulation element from the SARS-CoV-2 RNA genome. Nat. Struct. Mol. Biol. 2021, 28, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Manfredonia, I.; Nithin, C.; Ponce-Salvatierra, A.; Ghosh, P.; Wirecki, T.K.; Marinus, T.; Ogando, N.S.; Snijder, E.J.; van Hemert, M.J.; Bujnicki, J.M.; et al. Genome-wide mapping of SARS-CoV-2 RNA. Nucleic Acids Res. 2020, 48, 12436–12452. [Google Scholar] [CrossRef] [PubMed]
- Rangan, R.; Zheludev, I.N.; Hagey, R.J.; Pham, E.A.; Wayment-Steele, H.K.; Glenn, J.S.; Das, R. RNA genome conservation and secondary structure in SARS-CoV-2 and SARS-related viruses: A first look. RNA 2020, 26, 937–959. [Google Scholar] [CrossRef]
- Musalgaonkar, S.; Moomau, C.A.; Dinman, J.D. Ribosomes in the balance: Structural equilibrium ensures translational fidelity and proper gene expression. Nucleic Acids Res. 2014, 42, 13384–13392. [Google Scholar] [CrossRef] [Green Version]
- Caliskan, N.; Katunin, V.I.; Belardinelli, R.; Peske, F.; Rodnina, M.V. Programmed –1 frameshifting by kinetic partitioning during impeded translocation. Cell 2014, 157, 1619–1631. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Petrov, A.; Johansson, M.; Tsai, A.; O’Leary, S.E.; Puglisi, J.D. Dynamic pathways of −1 translational frameshifting. Nature 2014, 512, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Bao, C.; Loerch, S.; Ling, C.; Korostelev, A.A.; Grigorieff, N.; Ermolenko, D.N. mRNA stem-loops can pause the ribosome by hindering A-site tRNA binding. Elife 2020, 9, e55799. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, Y.-G.; Park, H.-J. Identification of RNA pseudoknot-binding ligand that inhibits the −1 ribosomal frameshifting of SARS-coronavirus by structure-based virtual screening. J. Am. Chem. Soc. 2011, 133, 10094–10100. [Google Scholar] [CrossRef]
- Ritchie, D.B.; Soong, J.; Sikkema, W.K.; Woodside, M.T. Anti-frameshifting ligand reduces the conformational plasticity of the SARS virus pseudoknot. J. Am. Chem. Soc. 2014, 136, 2196–2199. [Google Scholar] [CrossRef]
- Neupane, K.; Munshi, S.; Zhao, M.; Ritchie, D.B.; Ileperuma, S.M.; Woodside, M.T. Anti-frameshifting ligand active against SARS coronavirus-2 is resistant to natural mutations of the frameshift-stimulatory pseudoknot. J. Mol. Biol. 2020, 432, 5843–5847. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tao, H.; Shen, S.; Miao, Z.; Li, L.; Jia, Y.; Zhang, H.; Bai, X.; Fu, X. A drug screening toolkit based on the –1 ribosomal frameshifting of SARS-CoV-2. Heliyon 2020, 6, e04793. [Google Scholar] [CrossRef] [PubMed]
- Haniff, H.S.; Tong, Y.; Liu, X.; Chen, J.L.; Suresh, B.M.; Andrews, R.J.; Peterson, J.M.; O’Leary, C.A.; Benhamou, R.I.; Moss, W.N.; et al. Targeting the SARS-CoV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Cent. Sci. 2020, 6, 1713–1721. [Google Scholar] [CrossRef]
- Ahn, D.-G.; Lee, W.; Choi, J.-K.; Kim, S.-J.; Plant, E.P.; Almazan, F.; Taylor, D.R.; Enjuanes, L.; Oh, J.-W. Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antiviral Res. 2011, 91, 1–10. [Google Scholar] [CrossRef]
- Hu, H.-T.; Cho, C.-P.; Lin, Y.-H.; Chang, K.-Y. A general strategy to inhibiting viral −1 frameshifting based on upstream attenuation duplex formation. Nucleic Acids Res. 2016, 44, 256–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, D.-G.; Yoon, G.Y.; Lee, S.; Ku, K.B.; Kim, C.; Kim, K.-D.; Kwon, Y.-C.; Kim, G.-W.; Kim, B.-T.; Kim, S.-J. A Novel Frameshifting Inhibitor Having Antiviral Activity against Zoonotic Coronaviruses. Viruses 2021, 13, 1639. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource–improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef]
- Rodriguez, A.; Laio, A. Clustering by fast search and find of density peaks. Science 2014, 344, 1492–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grentzmann, G.; Ingram, J.A.; Kelly, P.J.; Gesteland, R.F.; Atkins, J.F. A dual-luciferase reporter system for studying recoding signals. RNA 1998, 4, 479–486. [Google Scholar] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kiso, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Imai, M.; Takeda, M.; Kinoshita, N.; Ohmagari, N.; Gohda, J.; Semba, K.; et al. The anticoagulant nafamostat potently inhibits SARS-CoV-2 S protein-mediated fusion in a cell fusion assay system and viral infection in vitro in a cell-type-dependent manner. Viruses 2020, 12, 629. [Google Scholar] [CrossRef]
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Nafamostat mesylate blocks activation of SARS-CoV-2, new treatment option for COVID-19. Antimicrob. Agents Chemother. 2020, 64, e00754-20. [Google Scholar] [CrossRef] [Green Version]
- Eggersmann, T.K.; Degenhardt, T.; Gluz, O.; Wuerstlein, R.; Harbeck, N. CDK4/6 inhibitors expand the therapeutic options in breast cancer: Palbociclib, ribociclib and abemaciclib. Biodrugs 2019, 33, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2021, 81, 317–331. [Google Scholar] [CrossRef]
- Tang, Y.Z.; Liu, Y.-H.; Chen, J.-X. Pleuromutilin and its derivatives-the lead compounds for novel antibiotics. Mini Rev. Med. Chem. 2012, 12, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Nixon, P.L.; Rangan, A.; Kim, Y.-G.; Rich, A.; Hoffman, D.W.; Hennig, M.; Giedroc, D.P. Solution structure of a luteoviral P1–P2 frameshifting mRNA pseudoknot. J. Mol. Biol. 2002, 322, 621–633. [Google Scholar] [CrossRef]
- Ritchie, D.B.; Cappellano, T.R.; Tittle, C.; Rezajooei, N.; Rouleau, L.; Sikkema, W.K.A.; Woodside, M.T. Conformational dynamics of the frameshift stimulatory structure in HIV-1. RNA 2017, 23, 1376–1384. [Google Scholar] [CrossRef]
- Gaudin, C.; Mazauric, M.-H.; Traïkia, M.; Guittet, E.; Yoshizawa, S.; Fourmy, D. Structure of the RNA signal essential for translational frameshifting in HIV-1. J. Mol. Biol. 2005, 349, 1024–1035. [Google Scholar] [CrossRef]
- Herold, J.; Raabe, T.; Schelle-Prinz, B.; Siddell, S. Nucleotide sequence of the human coronavirus 229E RNA polymerase locus. Virology 1993, 195, 680–691. [Google Scholar] [CrossRef]
- Brierley, I.; Boursnell, M.E.; Binns, M.M.; Bilimoria, B.; Blok, V.C.; Brown, T.D.; Inglis, S.C. An efficient ribosomal frame-shifting signal in the polymerase-encoding region of the coronavirus IBV. EMBO 1987, 6, 3779–3785. [Google Scholar] [CrossRef]
- Yamamoto, M.; Matsuyama, S.; Li, X.; Takeda, M.; Kawaguchi, Y.; Inoue, J.-I.; Matsuda, Z. Identification of nafamostat as a potent inhibitor of Middle East respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob. Agents Chemother. 2016, 60, 6532–6539. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, D.B.; Foster, D.A.; Woodside, M.T. Programmed −1 frameshifting efficiency correlates with RNA pseudoknot conformational plasticity, not resistance to mechanical unfolding. Proc. Natl. Acad. Sci. USA 2012, 109, 16167–16172. [Google Scholar] [CrossRef] [Green Version]
- Moomau, C.; Musalgaonkar, S.; Khan, Y.A.; Jones, J.E.; Dinman, J.D. Structural and functional characterization of programmed ribosomal frameshift signals in West Nile virus strains reveals high structural plasticity among cis-acting RNA elements. J. Biol. Chem. 2016, 291, 15788–15795. [Google Scholar] [CrossRef] [Green Version]
- Halma, M.T.; Ritchie, D.B.; Cappellano, T.R.; Neupane, K.; Woodside, M.T. Complex dynamics under tension in a high-efficiency frameshift stimulatory structure. Proc. Natl. Acad. Sci. USA 2019, 116, 19500–19505. [Google Scholar] [CrossRef]
- De Messieres, M.; Chang, J.-C.; Belew, A.T.; Meskauskas, A.; Dinman, J.D.; La Porta, A. Single-molecule measurements of the CCR5 mRNA unfolding pathways. Biophys. J. 2014, 106, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Halma, M.T.; Ritchie, D.B.; Woodside, M.T. Conformational Shannon entropy of mRNA structures from force spectroscopy measurements predicts the efficiency of −1 programmed ribosomal frameshift stimulation. Phys. Rev. Lett. 2021, 126, 038102. [Google Scholar] [CrossRef]
- Liu, T.; Kaplan, A.; Alexander, L.; Yan, S.; Wen, J.D.; Lancaster, L.; Wickersham, C.E.; Fredrick, K.; Noller, H.; Tinoco, I., Jr.; et al. Direct measurement of the mechanical work during translocation by the ribosome. Elife 2014, 3, e03406. [Google Scholar] [CrossRef]
- Yan, S.; Wen, J.-D.; Bustamante, C.; Tinoco, I., Jr. Ribosome excursions during mRNA translocation mediate broad branching of frameshift pathways. Cell 2015, 160, 870–881. [Google Scholar] [CrossRef] [Green Version]
- Neupane, K.; Zhao, M.; Lyons, A.; Munshi, S.; Ileperuma, S.M.; Ritchie, D.B.; Hoffer, N.Q.; Narayan, A.; Woodside, M.T. Structural dynamics of single SARS-CoV-2 pseudoknot molecules reveal topologically distinct conformers. Nat. Commun. 2021, 12, 4749. [Google Scholar] [CrossRef] [PubMed]
- Janssen, S.; Giegerich, R. The RNA shapes studio. Bioinformatics 2015, 31, 423–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Rastegari, B.; Condon, A.; Hoos, H.H. HotKnots: Heuristic prediction of RNA secondary structures including pseudoknots. RNA 2005, 11, 1494–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andronescu, M.S.; Pop, C.; Condon, A.E. Improved free energy parameters for RNA pseudoknotted secondary structure prediction. RNA 2010, 16, 26–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, L.E.; Shibata, Y.; Lukavsky, P.J. Rapid, nondenaturing RNA purification using weak anion-exchange fast performance liquid chromatography. RNA 2010, 16, 647–653. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Maurano, M.T.; Wang, H.; Qi, H.; Song, C.Z.; Navas, P.A.; Emery, D.W.; Stamatoyannopoulos, J.A.; Stamatoyannopoulos, G. Genomic discovery of potent chromatin insulators for human gene therapy. Nat. Biotechnol. 2015, 33, 198–203. [Google Scholar] [CrossRef]
- Cardno, T.S.; Poole, E.S.; Mathew, S.F.; Graves, R.; Tate, W.P.R. A homogeneous cell-based bicistronic fluorescence assay for high-throughput identification of drugs that perturb viral gene recoding and read-through of nonsense stop codons. RNA 2009, 15, 1614–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plant, E.P.; Dinman, J.D. Comparative study of the effects of heptameric slippery site composition on −1 frameshifting among different eukaryotic systems. RNA 2006, 12, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, A.; van Duin, J.; Pleij, C.W. Differential response to frameshift signals in eukaryotic and prokaryotic translational systems. Nucleic Acids Res. 1993, 21, 401–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, D.; Kang, H. Prokaryotic and eukaryotic translational machineries respond differently to the frameshifting RNA signal from plant or animal virus. Virus Res. 2003, 92, 165–170. [Google Scholar] [CrossRef]





{kind=link}
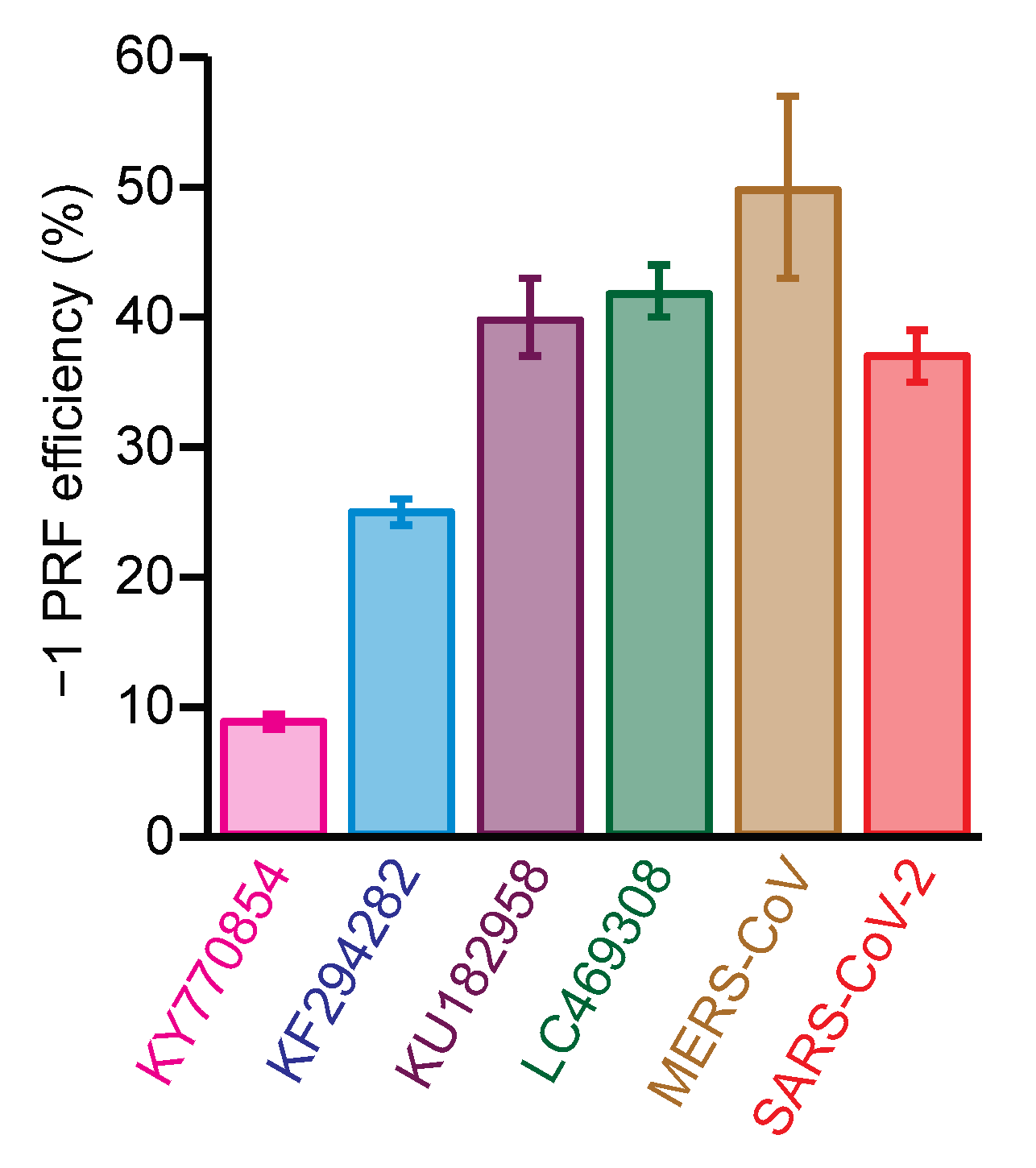{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 (μM) |
|---|---|
| nafamostat | 0.5 ± 0.4 |
| abemaciclib | 0.6 ± 0.2 |
| palbociclib | 0.6 ± 0.3 |
| valnemulin | 0.04 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munshi, S.; Neupane, K.; Ileperuma, S.M.; Halma, M.T.J.; Kelly, J.A.; Halpern, C.F.; Dinman, J.D.; Loerch, S.; Woodside, M.T. Identifying Inhibitors of −1 Programmed Ribosomal Frameshifting in a Broad Spectrum of Coronaviruses. Viruses 2022, 14, 177. https://doi.org/10.3390/v14020177
Munshi S, Neupane K, Ileperuma SM, Halma MTJ, Kelly JA, Halpern CF, Dinman JD, Loerch S, Woodside MT. Identifying Inhibitors of −1 Programmed Ribosomal Frameshifting in a Broad Spectrum of Coronaviruses. Viruses. 2022; 14(2):177. https://doi.org/10.3390/v14020177
Chicago/Turabian StyleMunshi, Sneha, Krishna Neupane, Sandaru M. Ileperuma, Matthew T. J. Halma, Jamie A. Kelly, Clarissa F. Halpern, Jonathan D. Dinman, Sarah Loerch, and Michael T. Woodside. 2022. "Identifying Inhibitors of −1 Programmed Ribosomal Frameshifting in a Broad Spectrum of Coronaviruses" Viruses 14, no. 2: 177. https://doi.org/10.3390/v14020177
APA StyleMunshi, S., Neupane, K., Ileperuma, S. M., Halma, M. T. J., Kelly, J. A., Halpern, C. F., Dinman, J. D., Loerch, S., & Woodside, M. T. (2022). Identifying Inhibitors of −1 Programmed Ribosomal Frameshifting in a Broad Spectrum of Coronaviruses. Viruses, 14(2), 177. https://doi.org/10.3390/v14020177