
Bacteriophages Roam the Wheat Phyllosphere

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. DNA Extraction and Amplification
2.3. Library Preparation for Viral and Bacterial Samples for Sequencing
2.4. Quality Control and Assembly
2.5. Viral Identification and Species Level Clustering
2.6. Microbial Fraction Profiling and Host Assignment
2.7. vOTU Taxonomy Assignment
2.8. Relative Abundances and Average Read Depths of the Viral Populations
2.9. Viral Particle Estimation
2.10. Manual Recovery of Two Complete Viral Genomes
2.11. Comparative Genomics of APSE Variants
3. Results
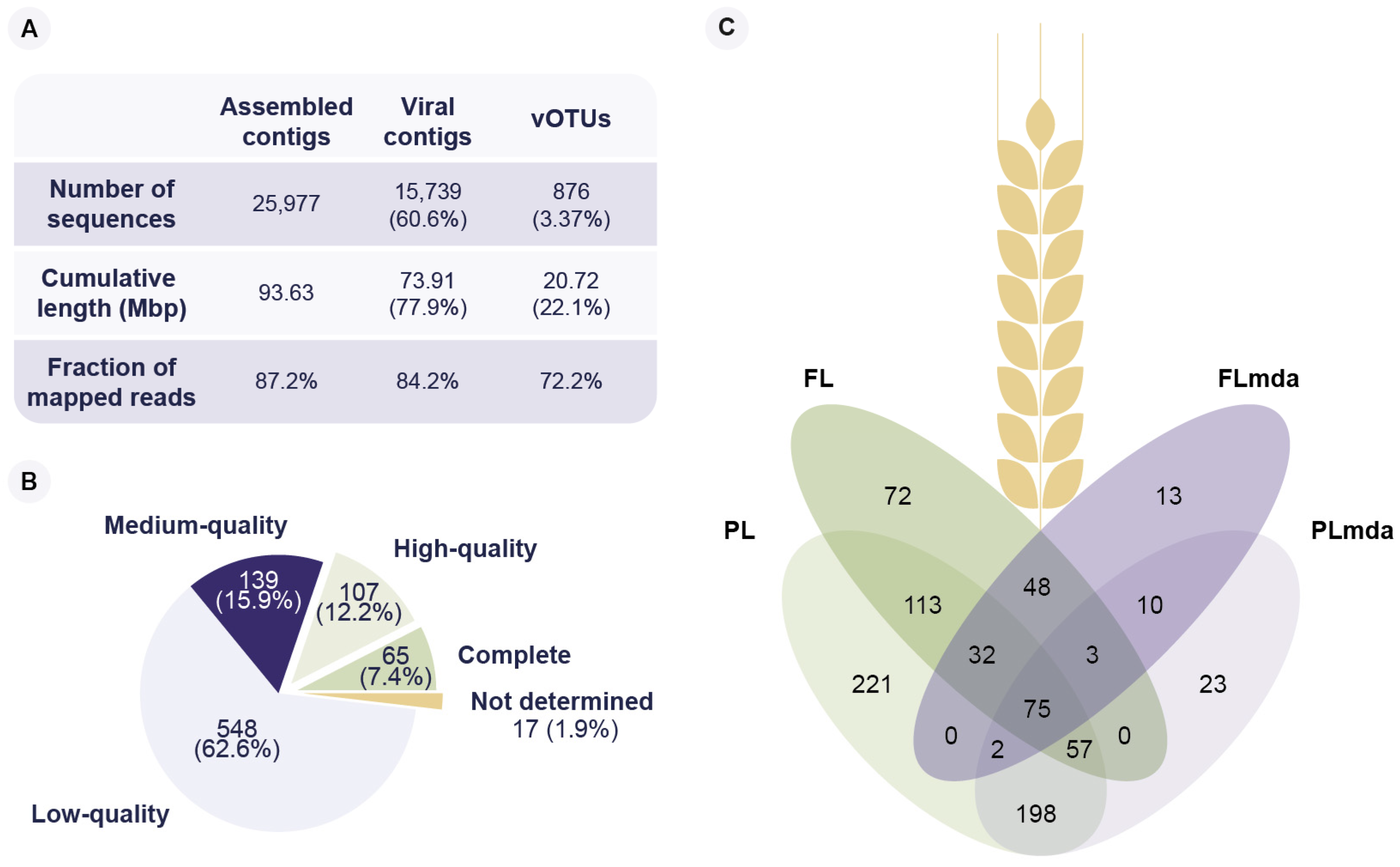
3.1. Diverse Phyllosphere Associated Bacteriophage Communities Can Be Successfully Recovered Using Viral Metagenomics
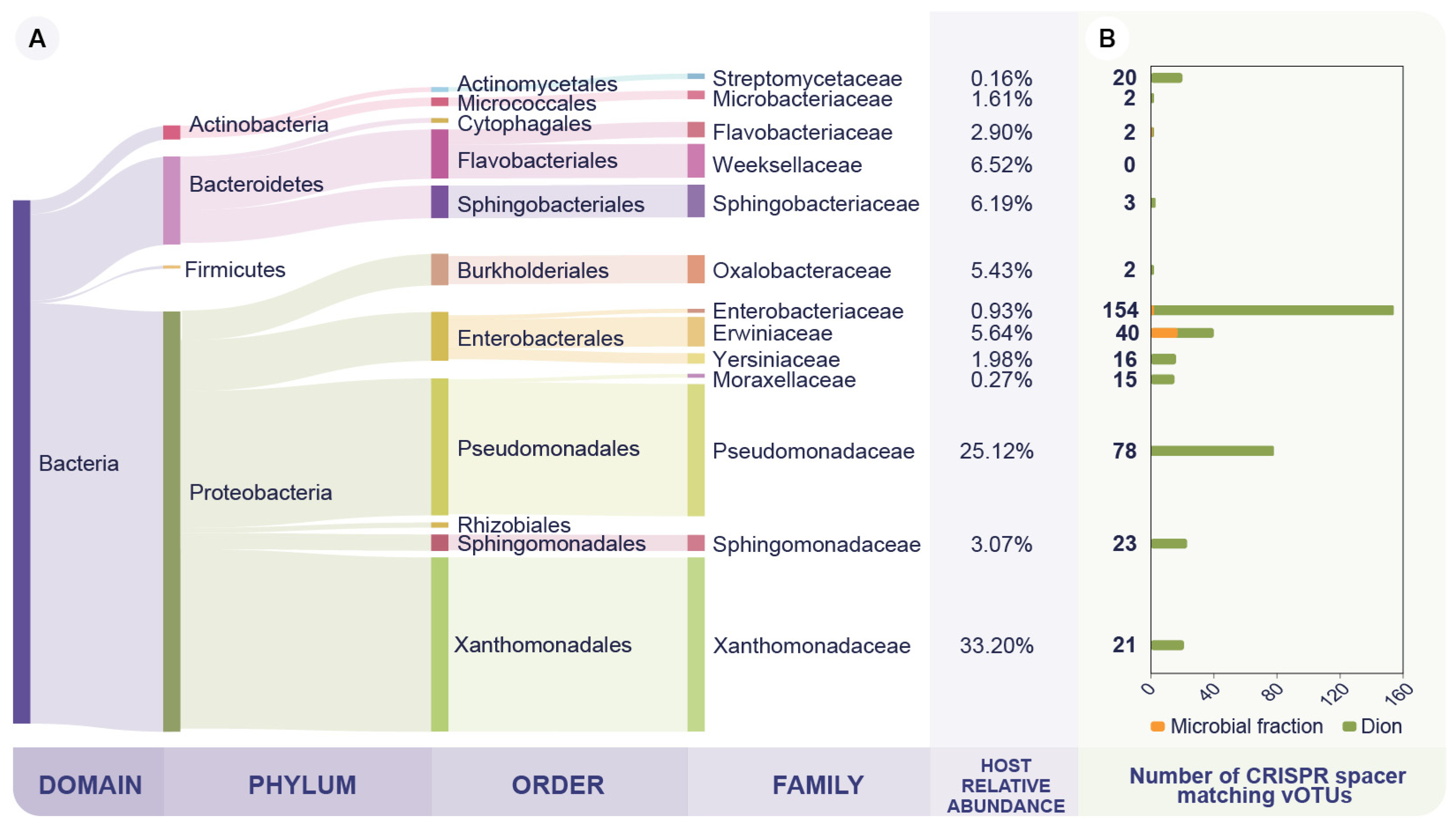
3.2. The Wheat Phyllosphere Microbial Fraction Is Dominated by Members of the Pseudomonadaceae Family
3.3. The Phyllosphere Associated Viral Community Is Predicted to Infect the Dominant Bacterial Families on the Microbial Fraction
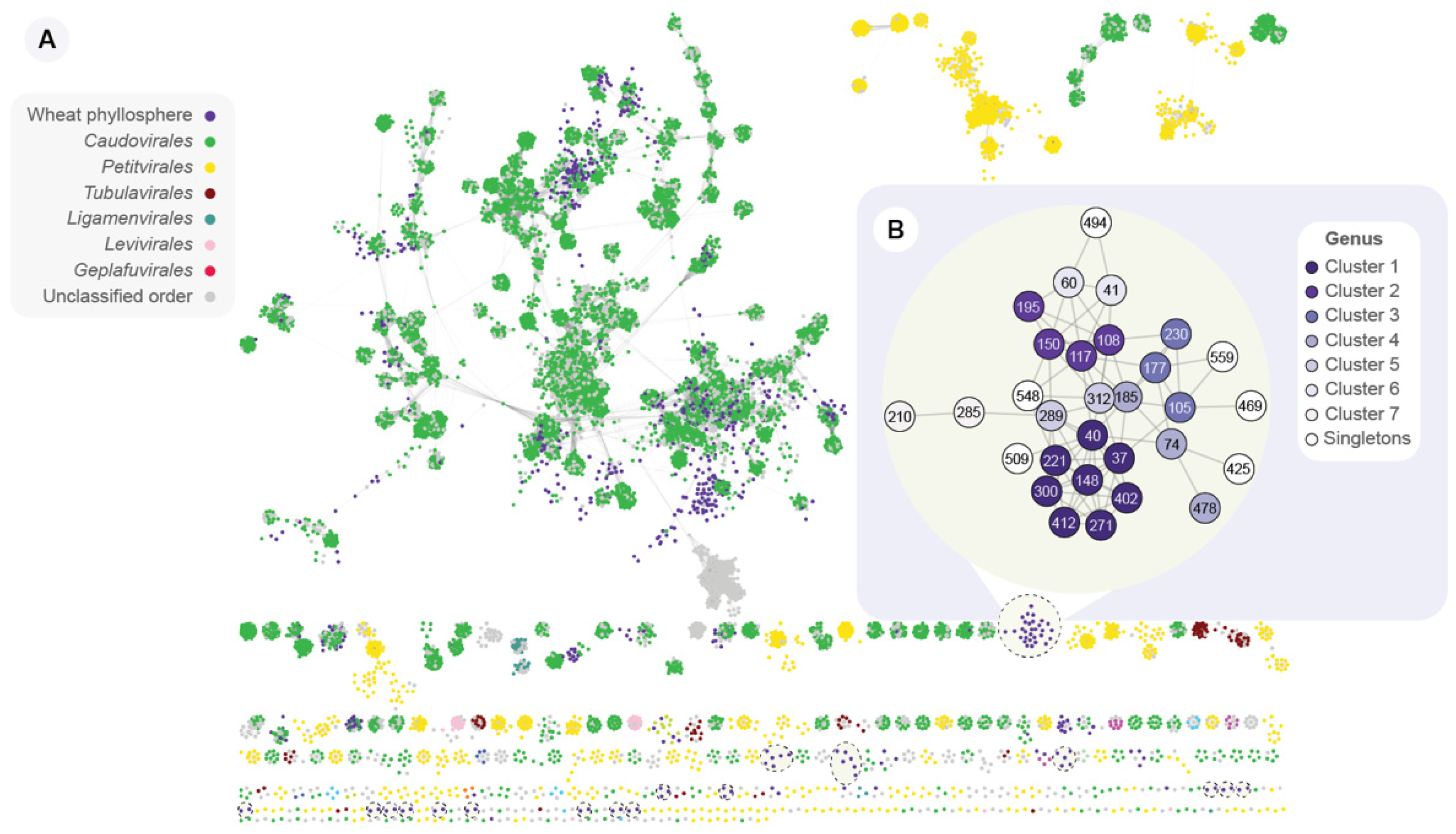
3.4. The Wheat Phyllosphere Harbors a Distinct Bacteriophage Community
3.5. The Top Rank-Abundant Phages Are Distinct on Each Leaf Type
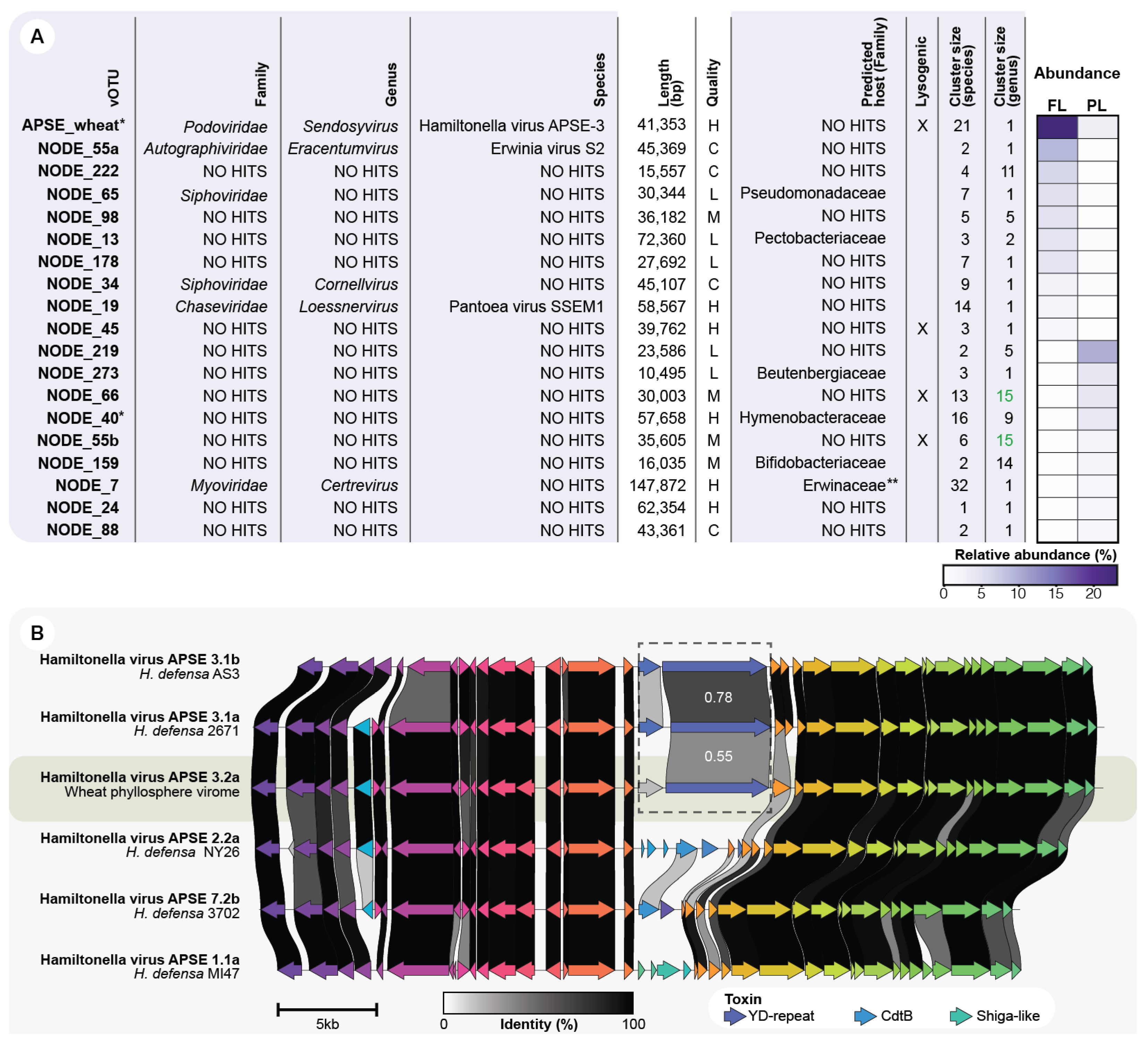
3.6. APSE Is the Most Abundant vOTU on the Phyllosphere and Carries a Predicted Insecticidal Toxin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B. The phyllosphere. Curr. Biol. 2020, 30, R1143–R1146. [Google Scholar] [CrossRef] [PubMed]
- Lindow, S.E.; Brandl, M.T. Microbiology of the Phyllosphere. Appl. Environ. Microbiol. 2003, 69, 1875–1883. [Google Scholar] [CrossRef] [Green Version]
- Rossmann, M.; Sarango-Flores, S.W.; Chiaramonte, J.B.; Kmit, M.C.P.; Mendes, R. Plant Microbiome: Composition and Functions in Plant Compartments. In The Brazilian Microbiome; Springer: Cham, Switzerland, 2017; pp. 7–20. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; van Themaat, E.V.L.; Schulze-Lefert, P. Structure and Functions of the Bacterial Microbiota of Plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.A.; Rothballer, M.; Chowdhury, S.P.; Nussbaumer, T.; Gutjahr, C.; Falter-Braun, P. Systems Biology of Plant-Microbiome Interactions. Mol. Plant 2019, 12, 804–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, J.; Wang, B.; Yoshikuni, Y. Microbiome Engineering: Synthetic Biology of Plant-Associated Microbiomes in Sustainable Agriculture. Trends Biotechnol. 2021, 39, 244–261. [Google Scholar] [CrossRef]
- Pratama, A.A.; Van Elsas, J.D. Gene mobility in microbiomes of the mycosphere and mycorrhizosphere—Role of plasmids and bacteriophages. FEMS Microbiol. Ecol. 2019, 95, fiz053. [Google Scholar] [CrossRef]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Sutton, T.D.S.; Hill, C. Gut Bacteriophage: Current Understanding and Challenges. Front. Endocrinol. 2019, 10, 784. [Google Scholar] [CrossRef]
- Von Wintersdorff, C.J.H.; Penders, J.; Van Niekerk, J.M.; Mills, N.D.; Majumder, S.; Van Alphen, L.B.; Savelkoul, P.H.M.; Wolffs, P.F.G. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 2016, 7, 173. [Google Scholar] [CrossRef] [Green Version]
- Breitbart, M. Marine Viruses: Truth or Dare. Annu. Rev. Mar. Sci. 2012, 4, 425–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morella, N.M.; Gomez, A.L.; Wang, G.; Leung, M.S.; Koskella, B. The impact of bacteriophages on phyllosphere bacterial abundance and composition. Mol. Ecol. 2018, 27, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Alcalá-Briseño, R.I.; Casarrubias-Castillo, K.; López-Ley, D.; Garrett, K.A.; Silva-Rosales, L. Network Analysis of the Papaya Orchard Virome from Two Agroecological Regions of Chiapas, Mexico. mSystems 2020, 5, e00423-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, Y.; Lian, S.; Chu, H.; Cho, J.K.; Yoo, S.-H.; Choi, H.; Yoon, J.-Y.; Choi, S.-K.; Lee, B.C.; Cho, W.K. Peach RNA viromes in six different peach cultivars. Sci. Rep. 2018, 8, 1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, P.L.C.; Badotti, F.; Oliveira, T.; Fonseca, A.; Vaz, A.B.M.; Tomé, L.M.R.; Abrahão, J.S.; Marques, J.T.; Trindade, G.S.; Chaverri, P.; et al. Virome analyses of Hevea brasiliensis using small RNA deep sequencing and PCR techniques reveal the presence of a potential new virus. Virol. J. 2018, 15, 184. [Google Scholar] [CrossRef] [PubMed]
- Peracchio, C.; Forgia, M.; Chiapello, M.; Vallino, M.; Turina, M.; Ciuffo, M. A complex virome including two distinct emaraviruses associated with virus-like symptoms in Camellia japonica. Virus Res. 2020, 286, 197964. [Google Scholar] [CrossRef]
- Redila, C.D.; Prakash, V.; Nouri, S. Metagenomics Analysis of the Wheat Virome Identifies Novel Plant and Fungal-Associated Viral Sequences. Viruses 2021, 13, 2457. [Google Scholar] [CrossRef]
- Akinyemi, I.A.; Wang, F.; Zhou, B.; Qi, S.; Wu, Q. Ecogenomic survey of plant viruses infecting Tobacco by Next generation sequencing. Virol. J. 2016, 13, 181. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Li, H.; Zhou, C.; Yang, Y. Effects of Flag Leaf and Number of Vegetative Ramets on Sexual Reproductive Performance in the Clonal Grass Leymus chinensis. Front. Plant Sci. 2020, 11, 534278. [Google Scholar] [CrossRef]
- Ma, J.; Tu, Y.; Zhu, J.; Luo, W.; Liu, H.; Li, C.; Li, S.; Liu, J.; Ding, P.; Habib, A.; et al. Flag leaf size and posture of bread wheat: Genetic dissection, QTL validation and their relationships with yield-related traits. Theor. Appl. Genet. 2020, 133, 297–315. [Google Scholar] [CrossRef]
- Waldren, R.P.; Flowerday, A.D. Growth Stages and Distribution of Dry Matter, N, P, and K in Winter Wheat. Agron. J. 1979, 71, 391–397. [Google Scholar] [CrossRef]
- Alanin, K.; Junco, L.; Jørgensen, J.; Nielsen, T.; Rasmussen, M.; Kot, W.; Hansen, L. Metaviromes Reveal the Dynamics of Pseudomonas Host-Specific Phages Cultured and Uncultured by Plaque Assay. Viruses 2021, 13, 959. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.; Krueger, F.; Seconds-Pichon, A.; Biggins, F.; Wingett, S. FastQC. A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2011. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Genome analysis Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef] [PubMed]
- Durai, D.A.; Schulz, M.H. Improving in-silico normalization using read weights. Sci. Rep. 2019, 9, 5133. [Google Scholar] [CrossRef]
- Rose, R.; Constantinides, B.; Tapinos, A.; Robertson, D.L.; Prosperi, M. Challenges in the analysis of viral metagenomes. Virus Evol. 2016, 2, vew022. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Roux, S.; Emerson, J.B.; Eloe-Fadrosh, E.A.; Sullivan, M.B. Benchmarking viromics: An in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 2017, 5, e3817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitwieser, F.P.; Salzberg, S.L. Pavian: Interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 2020, 36, 1303–1304. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Dion, M.B.; Plante, P.-L.; Zufferey, E.; Shah, S.A.; Corbeil, J.; Moineau, S. Streamlining CRISPR spacer-based bacterial host predictions to decipher the viral dark matter. Nucleic Acids Res. 2021, 49, 3127–3138. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Mirdita, M.; Karin, E.L.; Norroy, C.; Galiez, C.; Söding, J. SpacePHARER: Sensitive identification of phages from CRISPR spacers in prokaryotic hosts. Bioinformatics 2021, 37, 3364–3366. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI Viral Genomes Resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Páez-Espino, D.; Chen, I.M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Reddy, T.B.K.; Nayfach, S.; Schulz, F.; Call, L.; et al. IMG/VR v3: An integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2021, 49, D764–D775. [Google Scholar] [CrossRef] [PubMed]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouïl, J.; Jousselin, E.; D’Acier, A.C.; Cruaud, C.; Manzano-Marín, A. The Protector within: Comparative Genomics of APSE Phages across Aphids Reveals Rampant Recombination and Diverse Toxin Arsenals. Genome Biol. Evol. 2020, 12, 878–889. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Kim, K.-H.; Bae, J.-W. Amplification Methods Bias Metagenomic Libraries of Uncultured Single-Stranded and Double-Stranded DNA Viruses. Appl. Environ. Microbiol. 2011, 77, 7663–7668. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Bolduc, B.; Roux, S.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef]
- D’Humières, C.; Touchon, M.; Dion, S.; Cury, J.; Ghozlane, A.; Garcia-Garcera, M.; Bouchier, C.; Ma, L.; Denamur, E.; Rocha, E.P. A simple, reproducible and cost-effective procedure to analyse gut phageome: From phage isolation to bioinformatic approach. Sci. Rep. 2019, 9, 11331. [Google Scholar] [CrossRef] [Green Version]
- Batinovic, S.; Wassef, F.; Knowler, S.A.; Rice, D.T.; Stanton, C.R.; Rose, J.; Tucci, J.; Nittami, T.; Vinh, A.; Drummond, G.R.; et al. Bacteriophages in Natural and Artificial Environments. Pathogens 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gdanetz, K.; Trail, F. The Wheat Microbiome under Four Management Strategies, and Potential for Endophytes in Disease Protection. Phytobiomes, J. 2017, 1, 158–168. [Google Scholar] [CrossRef] [Green Version]
- Mukhtar, S.; Ishaq, A.; Hassan, S.; Mehnaz, S.; Mirza, M.S.; Malik, K.A. Comparison of Microbial Communities Associated with Halophyte (Salsola stocksii) and Non-Halophyte (Triticum aestivum) Using Culture-Independent Approaches. Pol. J. Microbiol. 2017, 66, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.; Rodriguez-Brito, B.; Bangor, D.; McNairnie, P.; Breitbart, M.; Salamon, P.; Felts, B.; Nulton, J.; Mahaffy, J.; Rohwer, F. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinform. 2005, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.-H.; Chang, H.-W.; Nam, Y.-D.; Roh, S.W.; Kim, M.-S.; Sung, Y.; Jeon, C.O.; Oh, H.-M.; Bae, J.-W. Amplification of Uncultured Single-Stranded DNA Viruses from Rice Paddy Soil. Appl. Environ. Microbiol. 2008, 74, 5975–5985. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, S.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef]
- Iriarte, F.B.; Balogh, B.; Momol, M.T.; Smith, L.M.; Wilson, M.; Jones, J.B. Factors Affecting Survival of Bacteriophage on Tomato Leaf Surfaces. Appl. Environ. Microbiol. 2007, 73, 1704–1711. [Google Scholar] [CrossRef] [Green Version]
- Knowles, B.; Silveira, C.; Bailey, B.A.; Barott, K.; Cantu, V.A.; Cobián-Güemes, A.G.; Coutinho, F.; Dinsdale, E.; Felts, B.; Furby, K.A.; et al. Lytic to temperate switching of viral communities. Nature 2016, 531, 466–470. [Google Scholar] [CrossRef]
- Degnan, P.H.; Yu, Y.; Sisneros, N.; Wing, R.; Moran, N.A. Hamiltonella defensa, genome evolution of protective bacterial endosymbiont from pathogenic ancestors. Proc. Natl. Acad. Sci. USA 2009, 106, 9063–9068. [Google Scholar] [CrossRef] [Green Version]
- Weldon, S.R.; Strand, M.R.; Oliver, K.M. Phage loss and the breakdown of a defensive symbiosis in aphids. Proc. R. Soc. B Boil. Sci. 2013, 280, 20122103. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, B.; Biosca, E.G. Bacteriophage-Based Bacterial Wilt Biocontrol for an Environmentally Sustainable Agriculture. Front. Plant Sci. 2017, 8, 1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, A.; Fujisawa, M.; Hamasaki, R.; Kawasaki, T.; Fujie, M.; Yamada, T. Biocontrol of Ralstonia solanacearum by Treatment with Lytic Bacteriophages. Appl. Environ. Microbiol. 2011, 77, 4155–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forero-Junco, L.M.; Alanin, K.W.S.; Djurhuus, A.M.; Kot, W.; Gobbi, A.; Hansen, L.H. Bacteriophages Roam the Wheat Phyllosphere. Viruses 2022, 14, 244. https://doi.org/10.3390/v14020244
Forero-Junco LM, Alanin KWS, Djurhuus AM, Kot W, Gobbi A, Hansen LH. Bacteriophages Roam the Wheat Phyllosphere. Viruses. 2022; 14(2):244. https://doi.org/10.3390/v14020244
Chicago/Turabian StyleForero-Junco, Laura Milena, Katrine Wacenius Skov Alanin, Amaru Miranda Djurhuus, Witold Kot, Alex Gobbi, and Lars Hestbjerg Hansen. 2022. "Bacteriophages Roam the Wheat Phyllosphere" Viruses 14, no. 2: 244. https://doi.org/10.3390/v14020244
APA StyleForero-Junco, L. M., Alanin, K. W. S., Djurhuus, A. M., Kot, W., Gobbi, A., & Hansen, L. H. (2022). Bacteriophages Roam the Wheat Phyllosphere. Viruses, 14(2), 244. https://doi.org/10.3390/v14020244