
Viruses Infecting Greenhood Orchids (Pterostylidinae) in Eastern Australia


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Surveys and Plant Samples
2.2. High-Throughput Sequencing and Sequence Assembly
2.3. Rapid Amplification of cDNA Ends (RACE)
2.4. Phylogenetic Analyses and Pairwise Sequence Comparisons
3. Results
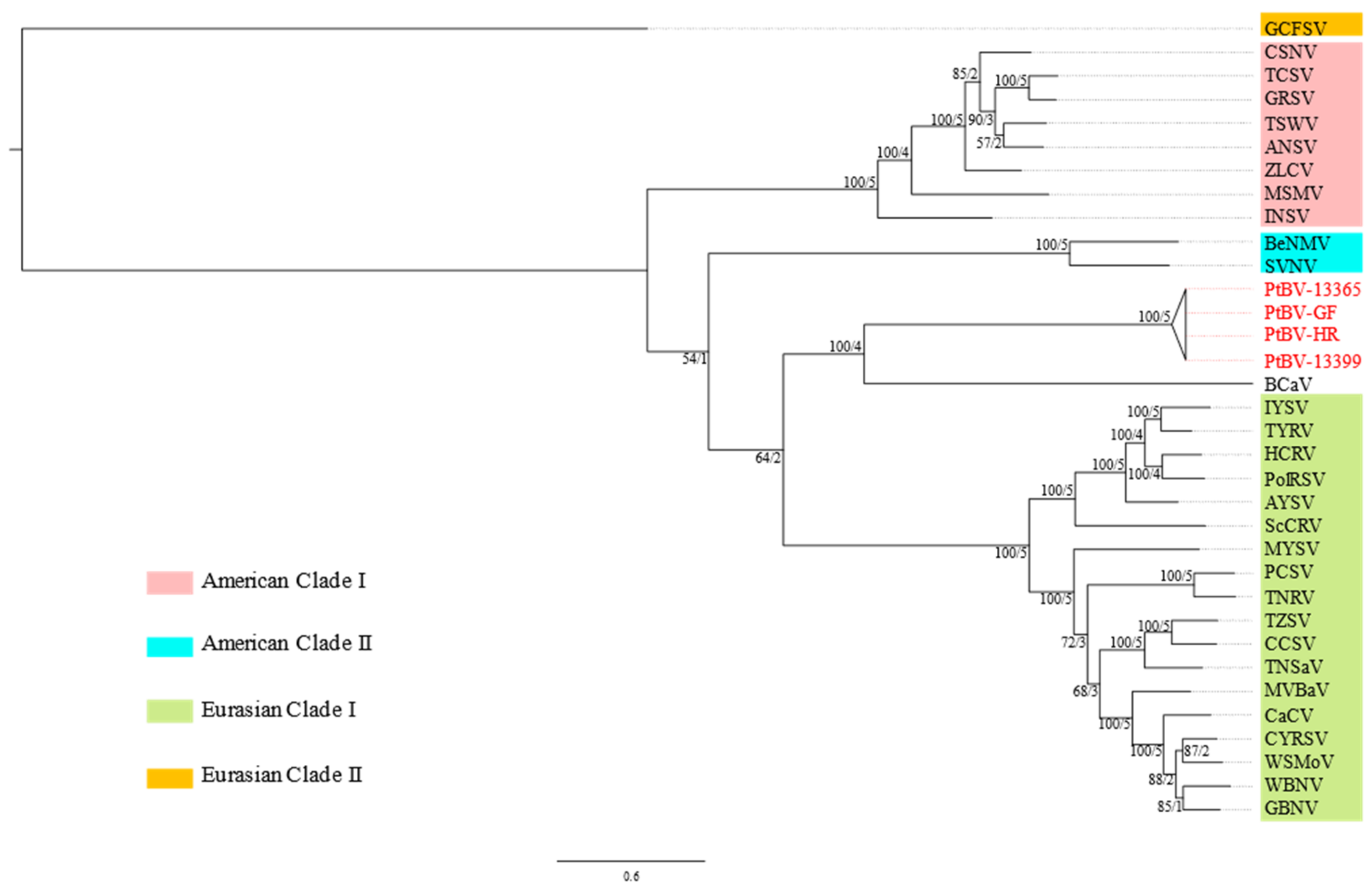
3.1. Molecular Characterisation of PtBV
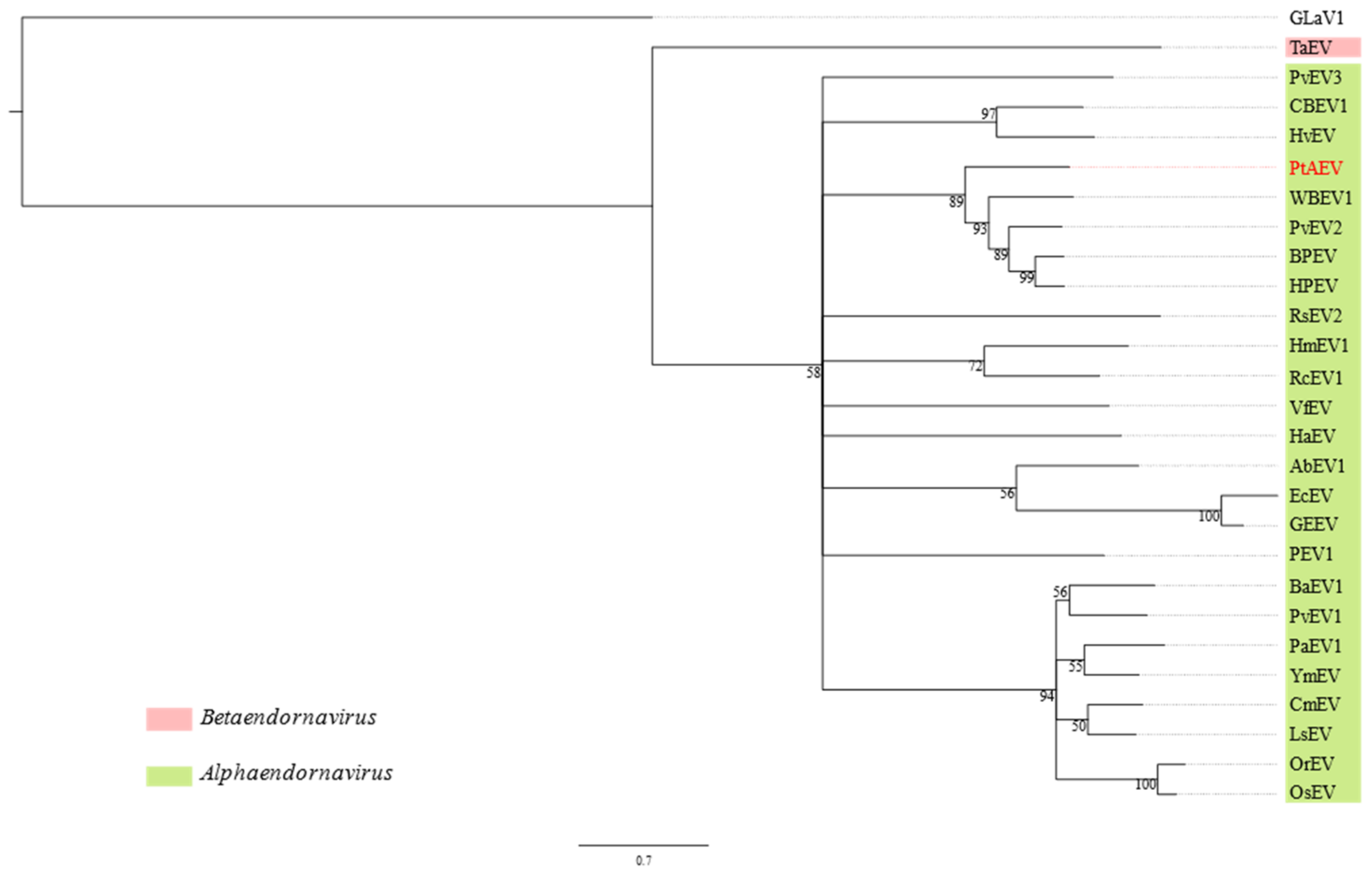
3.2. Pterostylis Alphaendornavirus
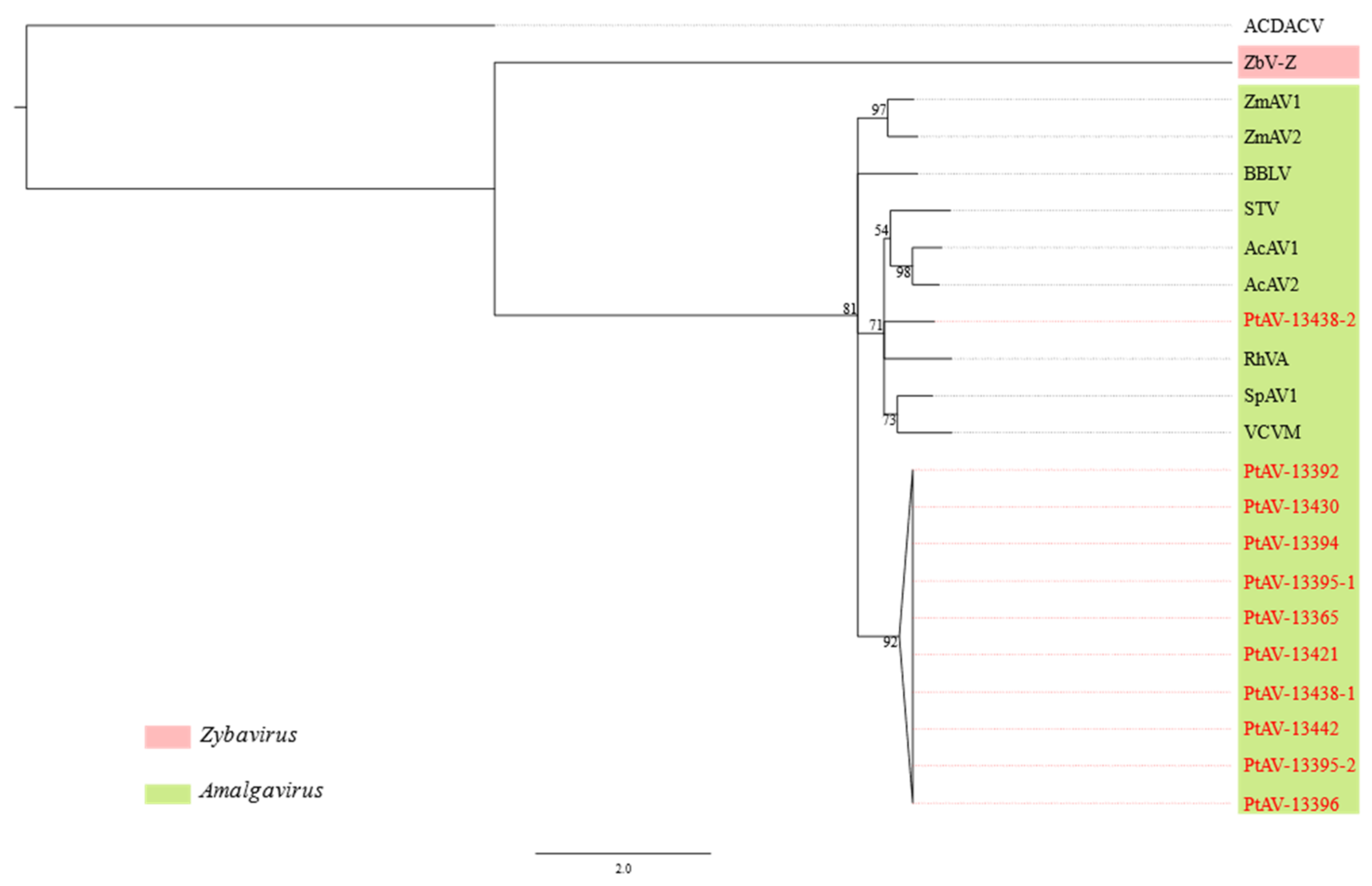
3.3. Pterostylis Amalgaviruses
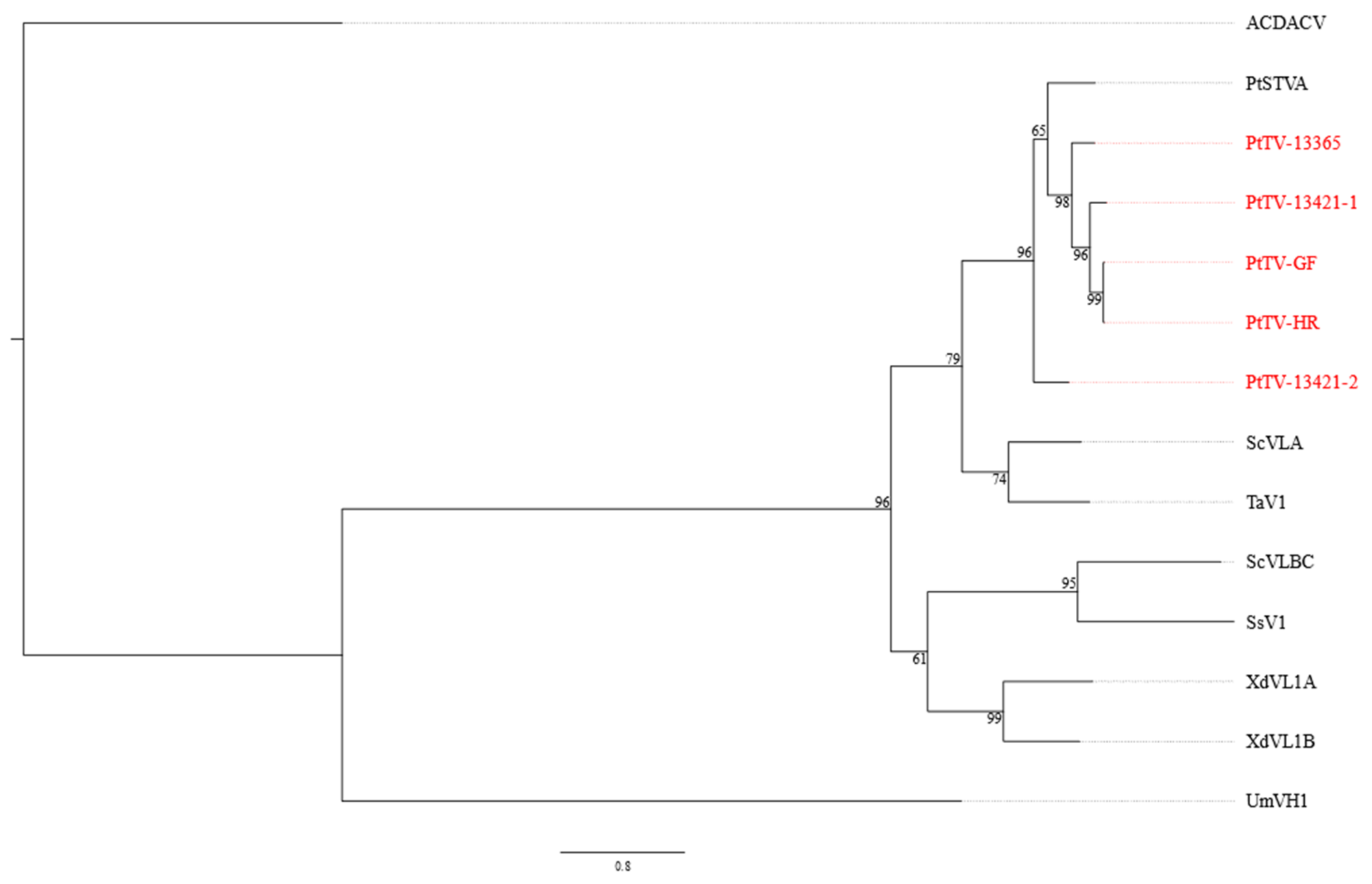
3.4. Pterostylis Totiviruses
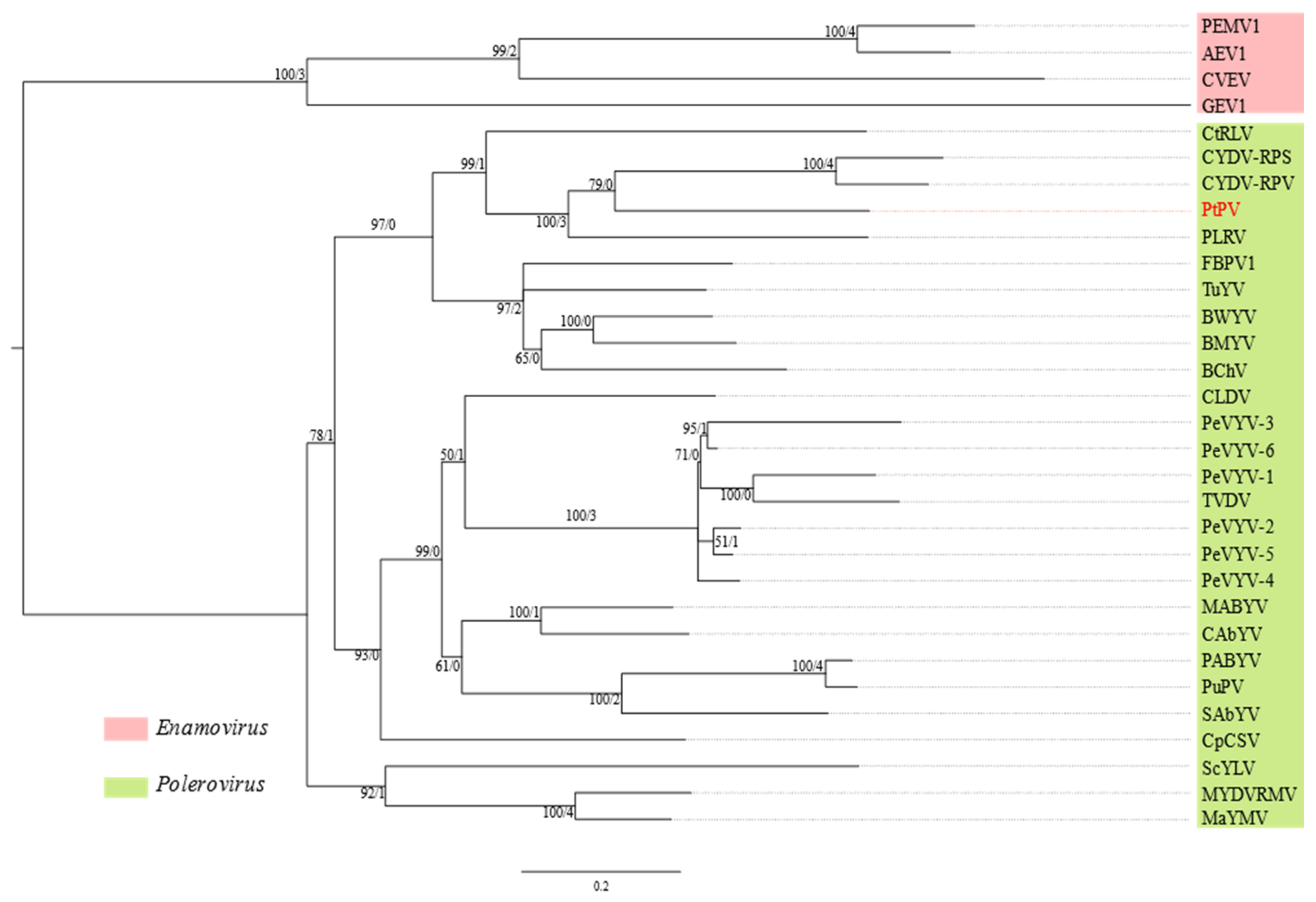
3.5. Pterostylis Polerovirus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webster, C.G.; Coutts, B.A.; Jones, R.A.C.; Jones, M.G.K.; Wylie, S.J. Virus impact at the interface of an ancient ecosystem and a recent agroecosystem: Studies on three legume-infecting potyviruses in the Southwest Australian floristic region. Plant Pathol. 2007, 56, 729–742. [Google Scholar] [CrossRef] [Green Version]
- García-Arenal, F.; Zerbini, F.M. Life on the edge: Geminiviruses at the Interface between Crops and Wild Plant Hosts. Annu. Rev. Virol. 2019, 6, 411–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdon, J.J.; Thrall, P.H.; Ericson, L. The Current and Future Dynamics of Disease in Plant Communities. Annu. Rev. Phytopathol. 2006, 44, 19–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastogeer, K.M.G.; Li, H.; Sivasithamparam, K.; Jones, M.G.K.; Wylie, S.J. Fungal endophytes and a virus confer drought tolerance to Nicotiana benthamiana plants through modulating osmolytes, antioxidant enzymes and expression of host drought responsive genes. Environ. Exp. Bot. 2018, 149, 95–108. [Google Scholar] [CrossRef] [Green Version]
- WFO: World Flora Online. 2021. Available online: http://www.worldfloraonline.org (accessed on 16 November 2021).
- Givnish, T.J.; Spalink, D.; Ames, M.; Lyon, S.P.; Hunter, S.J.; Zuluaga, A.; Doucette, A.; Caro, G.G.; McDaniel, J.; Clements, M.A. Orchid historical biogeography, diversification, Antarctica and the paradox of orchid dispersal. J. Biogeogr. 2016, 43, 1905–1916. [Google Scholar] [CrossRef]
- Li, Y.-X.; Li, Z.-H.; Schuiteman, A.; Chase, M.W.; Li, J.-W.; Huang, W.-C.; Hidayat, A.; Wu, S.-S.; Jin, X.-H. Phylogenomics of Orchidaceae based on plastid and mitochondrial genomes. Mol. Phylogenetics Evol. 2019, 139, 106540. [Google Scholar] [CrossRef] [PubMed]
- Clements, M.A.; Otero, J.T.; Miller, J.T. Phylogenetic relationships in Pterostylidinae (Cranichideae: Orchidaceae): Combined evidence from nuclear ribomsomal and plastid DNA sequences. Aust. J. Bot. 2011, 59, 99–117. [Google Scholar] [CrossRef]
- Pridgeon, A.M.; Cribb, J.P.; Chase, W.M.; Rasmussen, F. Genera Orchidacearum; Oxford University Press: Oxford, UK, 1999–2014. [Google Scholar]
- Jones, D.L. A Complete Guide to Native Orchids of Australia, 3rd ed.; New Holland Publishers Frenchs Forest: Sydney, Australia, 2020. [Google Scholar]
- Warcup, J.H. Symbiotic germination of some Australian terrestrial orchids. New Phytol. 1973, 72, 387–392. [Google Scholar] [CrossRef]
- Otero, J.T.; Thrall, P.H.; Clements, M.; Burdon, J.J.; Miller, J.T. Codiversification of orchids (Pterostylidinae) and their associated mycorrhizal fungi. Aust. J. Bot. 2011, 59, 480–497. [Google Scholar] [CrossRef]
- Hayashi, T.; Bohman, B.; Scaffidi, A.; Peakall, R.; Flematti, G.R. An unusual tricosatriene is crucial for male fungus gnat attraction and exploitation by sexually deceptive Pterostylis orchids. Curr. Biol. 2021, 31, 1954–1961.e7. [Google Scholar] [CrossRef] [PubMed]
- Zettler, F.W.; Ko, N.J.; Wisler, G.C.; Elliott, M.S.; Wong, S.M. Viruses of orchids and their control. Plant Dis. 1990, 74, 621–626. [Google Scholar] [CrossRef]
- Gibbs, A.; Mackenzie, A.; Blanchfield, A.; Cross, P.; Wilson, C.; Elliot, K.; Nightingale, M.; Clements, M. Viruses of orchids in Australia: Their identification, biology and control. Aust. Orchid Rev. 2000, 65, 10–21. [Google Scholar]
- Ali, R.N.; Dann, A.L.; Cross, P.A.; Wilson, C.R. Multiplex RT-PCR detection of three common viruses infecting orchids. Arch. Virol. 2014, 159, 3095–3099. [Google Scholar] [CrossRef] [PubMed]
- Dietzgen, R.G.; Freitas-Astúa, J.; Chabi-Jesus, C.; Ramos-González, P.L.; Goodin, M.M.; Kondo, H.; Tassi, A.D.; Kitajima, E.W. Chapter Five - Dichorhaviruses in their Host Plants and Mite Vectors. In Advances in Virus Research; Palukaitis, P., Roossinck, M.J., Eds.; Academic Press: Maryland Heights, MO, USA, 2018; Volume 102, Chapter 5; pp. 119–148. [Google Scholar]
- Wylie, S.J.; Li, H.; Dixon, K.W.; Richards, H.; Jones, M.G.K. Exotic and indigenous viruses infect wild populations and captive collections of temperate terrestrial orchids (Diuris Species) in Australia. Virus Res. 2013, 171, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wylie, S.J.; Tan, A.J.Y.; Li, H.; Dixon, K.W.; Jones, M.G.K. Caladenia virus A, an unusual new member of the family Potyviridae from terrestrial orchids in Western Australia. Arch. Virol. 2012, 157, 2447–2452. [Google Scholar] [CrossRef] [PubMed]
- Wylie, S.J.; Li, H.; Jones, M.G.K. Donkey Orchid Symptomless Virus: A Viral ‘Platypus’ from Australian Terrestrial Orchids. PLoS ONE 2013, 8, e79587. [Google Scholar]
- Ong, J.W.L.; Li, H.; Sivasithamparam, K.; Dixon, K.W.; Jones, M.G.K.; Wylie, S.J. Novel Endorna-like viruses, including three with two open reading frames, challenge the membership criteria and taxonomy of the Endornaviridae. Virology 2016, 499, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Parrella, G.; Gognalons, P.; Gebre-Selassiè, K.; Vovlas, C.; Marchoux, G. An update of the host range of tomato spotted wilt virus. J. Plant Pathol. 2003, 85, 227–264. [Google Scholar]
- Persley, D.M.; Thomas, J.E.; Sharman, M. Tospoviruses—An Australian perspective. Australas. Plant Pathol. 2006, 35, 161–180. [Google Scholar] [CrossRef]
- Oliver, J.E.; Whitfield, A.E. The Genus Tospovirus: Emerging Bunyaviruses that Threaten Food Security. Annu. Rev. Virol. 2016, 3, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Kück, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. (Eds.) Family—Bunyaviridae. In Virus Taxonomy; Elsevier: San Diego, CA, USA, 2012; pp. 725–741. [Google Scholar]
- Valverde, R.A.; Khalifa, M.E.; Okada, R.; Fukuhara, T.; Sabanadzovic, S. ICTV Report Consortium ICTV Virus Taxonomy Profile: Endornaviridae. J. Gen. Virol. 2019, 100, 1204–1205. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. Plant viruses of the Amalgaviridae family evolved via recombination between viruses with double-stranded and negative-strand RNA genomes. Biol. Direct 2015, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Vainio, E.J.; Chiba, S.; Ghabrial, S.A.; Maiss, E.; Roossinck, M.; Sabanadzovic, S.; Suzuki, N.; Xie, J.; Nibert, M. ICTV Report Consortium ICTV Virus Taxonomy Profile: Partitiviridae. J. Gen. Virol. 2018, 99, 17–18. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. (Eds.) Family—Totiviridae. In Virus Taxonomy; Elsevier: San Diego, CA, USA, 2012; pp. 639–650. [Google Scholar]
- Morozov, S.Y.; Solovyev, A.G. Small hydrophobic viral proteins involved in intercellular movement of diverse plant virus genomes. AIMS Microbiol. 2020, 6, 305–329. [Google Scholar] [CrossRef] [PubMed]
- Sõmera, M.; Fargette, D.; Hébrard, E.; Sarmiento, C. ICTV Report Consortium ICTV Virus Taxonomy Profile: Solemoviridae 2021. J. Gen. Virol. 2021, 102, 001707. [Google Scholar] [CrossRef]
- Wang, F.; Dong, Q.; Gao, Z.; Zhou, B.; Bao, X. Characterisation of a novel alphaendornavirus isolated from balloon flower (Platycodon grandiflorus). Arch. Virol. 2020, 165, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Tzanetakis, I.E.; Sabanadzovic, S.; Valverde, R.A. Amalgaviruses (Amalgaviridae). In Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: Oxford, UK, 2021; pp. 154–157. [Google Scholar]
- Favre-Godal, Q.; Gourguillon, L.; Lordel-Madeleine, S.; Gindro, K.; Choisy, P. Orchids and their mycorrhizal fungi: An insufficiently explored relationship. Mycorrhiza 2020, 30, 5–22. [Google Scholar] [CrossRef]
- Ong, J.W.L.; Li, H.; Sivasithamparam, K.; Dixon, K.W.; Jones, M.G.K.; Wylie, S.J. Novel and divergent viruses associated with Australian orchid-fungus symbioses. Virus Res. 2018, 244, 276–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Impatiens Necrotic Spot Virus (INSV) in New South Wales (NSW). Available online: https://www.ippc.int/en/countries/australia/pestreports/2018/09/impatiens-necrotic-spot-virus-insv-in-new-south-wales-nsw/ (accessed on 3 November 2021).
- Janes, J.K.; Duretto, M.F. A new classification for subtribe Pterostylidinae (Orchidaceae), reaffirming Pterostylis in the broad sense. Aust. Syst. Bot. 2010, 23, 260–269. [Google Scholar] [CrossRef]
- Mound, L.A. Thysanoptera of the genus Dichromothrips on Old World Orchidaceae. Biol. J. Linn. Soc. 1976, 8, 245–265. [Google Scholar] [CrossRef]
- Mound, L.A.; Tree, D.J. Thysanoptera Australiensis—Thrips of Australia. 2020. Available online: https://keys.lucidcentral.org/keys/v3/thrips_australia/ (accessed on 18 October 2021).
- Read, D.A.; Roberts, R.; Thompson, G. Genomic characterization of two novel viruses infecting Barleria cristata L. from the genera Orthotospovirus and Polerovirus. Arch. Virol. 2021, 166, 2615–2618. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kanakala, S.; Lebedev, G.; Kontsedalov, S.; Silverman, D.; Alon, T.; Mor, N.; Sela, N.; Luria, N.; Dombrovsky, A.; et al. Transmission of a New Polerovirus Infecting Pepper by the Whitefly Bemisia tabaci. J. Virol. 2019, 93, e00488-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Ruiz, H.; Holste, N.M.; LaTourrette, K. Poleroviruses (Luteoviridae). In Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: Oxford, UK, 2021; pp. 594–602. [Google Scholar]
- Austin, A.D.; Yeates, D.K.; Cassis, G.; Fletcher, M.J.; La Salle, J.; Lawrence, J.F.; McQuillan, P.B.; Mound, L.A.; J Bickel, D.; Gullan, P.J. Insects ‘Down under’– Diversity, endemism and evolution of the Australian insect fauna: Examples from select orders. Aust. J. Entomol. 2004, 43, 216–234. [Google Scholar] [CrossRef]
- Schliephake, E.; Graichen, K.; Rabenstein, F. Investigations on the vector transmission of the Beet mild yellowing virus (BMYV) and the Turnip yellows virus (TuYV). Untersuchungen zur Vektorübertragung des Milden Rübenvergilbungsvirus (Beet mild yellowing virus) und des Wasserrübenvergilbungsvirus (Turnip yellows virus). Zeitschrift Pflanzenkrankheiten Pflanzenschutz J. Plant Dis. Prot. 2000, 107, 81–87. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Location | Host Species | Virus Genus | Sequence Notation | Sequence Depth * |
|---|---|---|---|---|---|
| 13365 | Black Mountain Lookout, ACT | Pterostylis nutans | Amalgavirus | PtAV-13365 | 77 |
| Orthotospovirus | PtBV-13365 | L: 80,318; M: 54,274; S: 47,192 | |||
| Polerovirus | PtPV | 1,790,464 | |||
| Totivirus | PtTV-13365 | 528 | |||
| 13392 | Camp Pincham track, Warrumbungle National Park, NSW | Pterostylis curta | Amalgavirus | PtAV-13392 | 262 |
| 13394 | Camp Pincham track, Warrumbungle National Park, NSW | Pterostylis curta | Amalgavirus | PtAV-13394 | 198 |
| 13395 | Camp Pincham track, Warrumbungle National Park, NSW | Pterostylis curta | Amalgavirus | PtAV-13395-1 | 95 |
| Amalgavirus | PtAV-13395-2 | 24 | |||
| 13396 | Camp Pincham track, Warrumbungle National Park, NSW | Pterostylis curta | Amalgavirus | PtAV-13396 | 53 |
| 13399 | Burbie Camp track, Warrumbungle National Park, NSW | Pterostylis curta | Orthotospovirus | PtBV-13399 | L:56,527; M: 87,708; S: 41,563 |
| 13402 | Timor Rock, Warrumbungle National Park, NSW | Pterostylis curta | - | - | - |
| 13421 | Governor Track, Mt Kaputar National Park, NSW | Pterostylis nutans | Amalgavirus | PtAV-13421 | 15 |
| Totivirus | PtTV-13421-1 | 665 | |||
| Totivirus | PtTV-13421-2 | 35 | |||
| 13430 | Wellington, Mt Arthur Reserve, NSW | Pterostylis curta | Amalgavirus | PtAV-13430 | 112 |
| 13438 | Grill Cave, Bungonia National Park, NSW | Pterostylis nutans | Amalgavirus | PtAV-13438-1 | 40 |
| Amalgavirus | PtAV-13438-2 | 15 | |||
| 13442 | Green track, Bungonia National Park, NSW | Pterostylis curta | Amalgavirus | PtAV-13442 | 97 |
| GF | Gibraltar Falls, Namadgi National Park, ACT | Pterostylis curta | Orthotospovirus | PtBV-GF | L: 12,876; M: 10,559; S: 3453 |
| Totivirus | PtTV-GF | 63 | |||
| HR | Hanging Rock, Tidbinbilla Nature Reserve, ACT | Pterostylis nutans | Alphaendornavirus | PtAEV | 68 |
| Orthotospovirus | PtBV-HR | L: 4026; M: 7010; S: 1195 | |||
| Totivirus | PtTV-HR | 36 | |||
| PI-1 | Pine Island Reserve, ACT | Oligochaetochilus hamatus | - | - | - |
| PI-2 | Pine Island Reserve, ACT | Pterostylis nutans | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, H.-Y.; Clements, M.A.; Mackenzie, A.M.; Dietzgen, R.G.; Thomas, J.E.; Geering, A.D.W. Viruses Infecting Greenhood Orchids (Pterostylidinae) in Eastern Australia. Viruses 2022, 14, 365. https://doi.org/10.3390/v14020365
Chao H-Y, Clements MA, Mackenzie AM, Dietzgen RG, Thomas JE, Geering ADW. Viruses Infecting Greenhood Orchids (Pterostylidinae) in Eastern Australia. Viruses. 2022; 14(2):365. https://doi.org/10.3390/v14020365
Chicago/Turabian StyleChao, Hsu-Yao, Mark A. Clements, Anne M. Mackenzie, Ralf G. Dietzgen, John E. Thomas, and Andrew D. W. Geering. 2022. "Viruses Infecting Greenhood Orchids (Pterostylidinae) in Eastern Australia" Viruses 14, no. 2: 365. https://doi.org/10.3390/v14020365
APA StyleChao, H. -Y., Clements, M. A., Mackenzie, A. M., Dietzgen, R. G., Thomas, J. E., & Geering, A. D. W. (2022). Viruses Infecting Greenhood Orchids (Pterostylidinae) in Eastern Australia. Viruses, 14(2), 365. https://doi.org/10.3390/v14020365