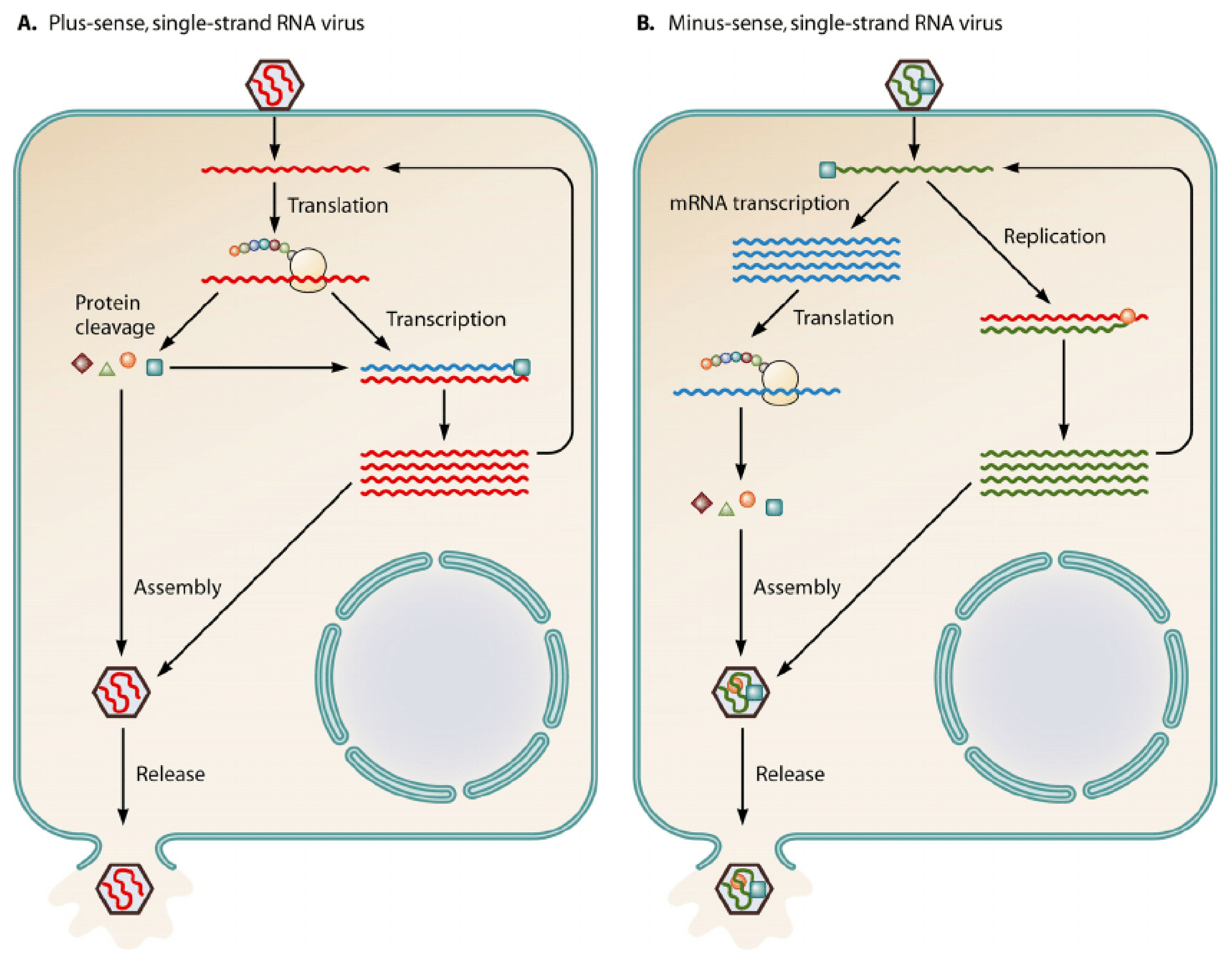
Figure 1.
The above diagram displays the notable differences between a (+)—sense single-stranded RNA virus and a (−)—sense single-stranded RNA virus [
2].
Figure 1.
The above diagram displays the notable differences between a (+)—sense single-stranded RNA virus and a (−)—sense single-stranded RNA virus [
2].
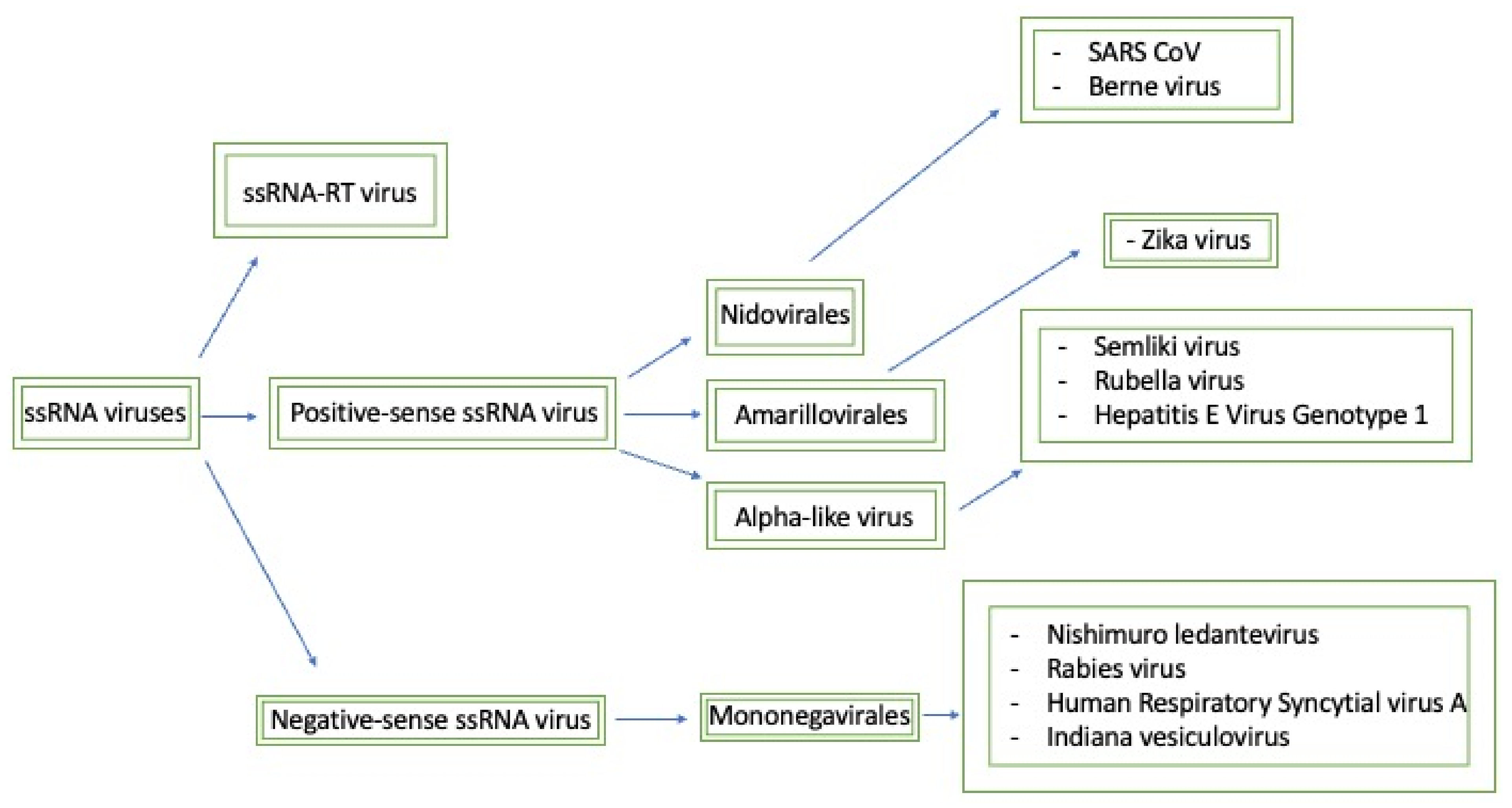
Figure 2.
This map seeks to briefly summarize the methyltransferases that will be discussed and how they relate to each other. Generally, the negative-sense ssRNA viruses have similar methyltransferase domains that are classified as mononegavirales-like methyltransferases. Furthermore, based on the sequence domain composition, positive-sense ssRNA viruses can be split into three separate families: nidovirales-like, NS5-like, and alpha-like methyltransferases.
Figure 2.
This map seeks to briefly summarize the methyltransferases that will be discussed and how they relate to each other. Generally, the negative-sense ssRNA viruses have similar methyltransferase domains that are classified as mononegavirales-like methyltransferases. Furthermore, based on the sequence domain composition, positive-sense ssRNA viruses can be split into three separate families: nidovirales-like, NS5-like, and alpha-like methyltransferases.
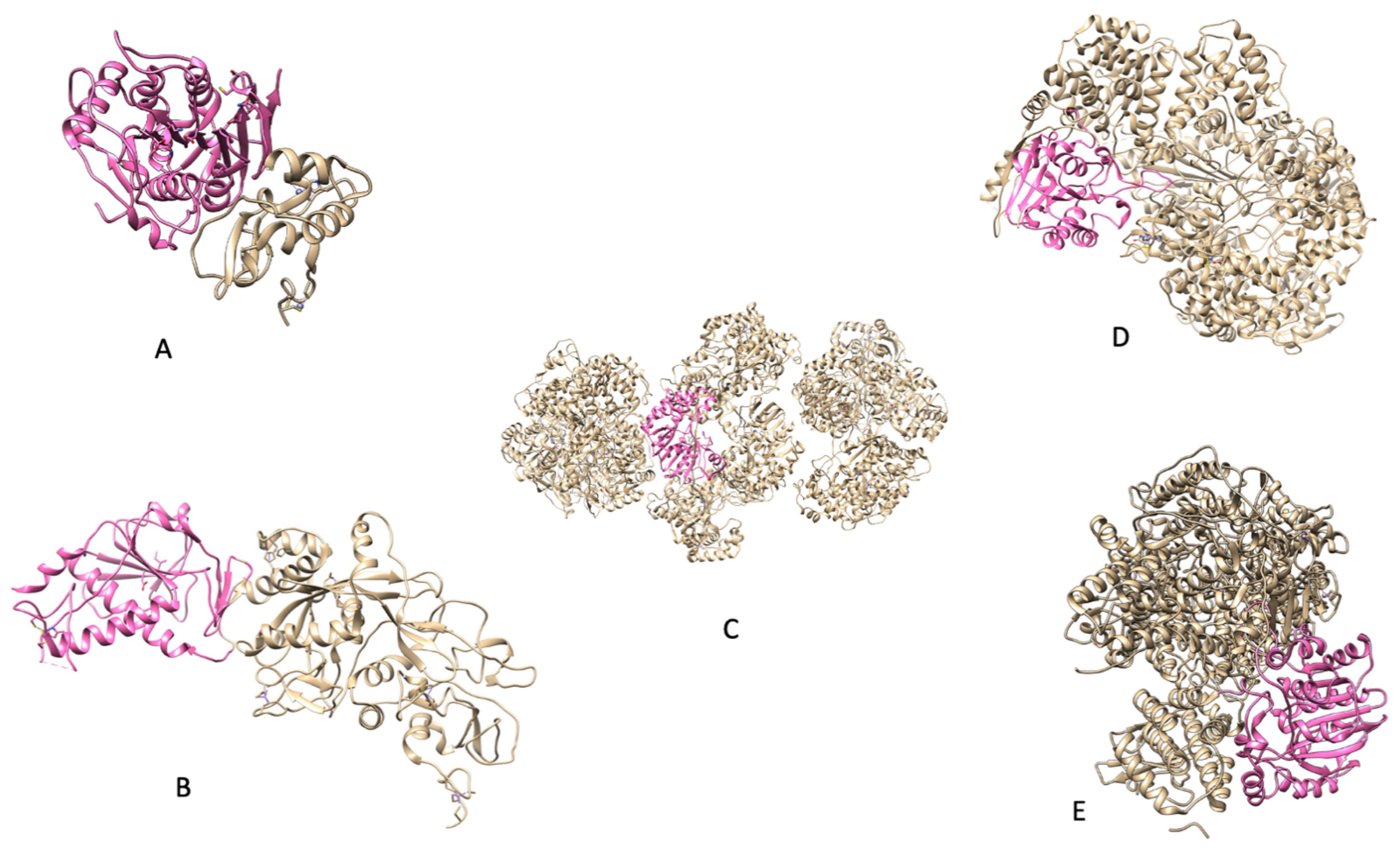
Figure 3.
(
A) PDB 3R24: NSP16 protein from
SARS CoV); (
B) (PDB 5C8U: NSP14 protein from
SARS CoV); (
C) (PDB 5M2X: NS5 protein from
Zika); (
D) [
8] (PDB 5A22: Protein L from
Indiana vesiculovirus); (
E) (PDB 6UEB: L protein from
Rabies virus).
Indiana vesiculovirus was used as its sequence is quite similar to that of other negative-strand ssRNA viruses and PDBs are not available for the viruses discussed.
Figure 3.
(
A) PDB 3R24: NSP16 protein from
SARS CoV); (
B) (PDB 5C8U: NSP14 protein from
SARS CoV); (
C) (PDB 5M2X: NS5 protein from
Zika); (
D) [
8] (PDB 5A22: Protein L from
Indiana vesiculovirus); (
E) (PDB 6UEB: L protein from
Rabies virus).
Indiana vesiculovirus was used as its sequence is quite similar to that of other negative-strand ssRNA viruses and PDBs are not available for the viruses discussed.
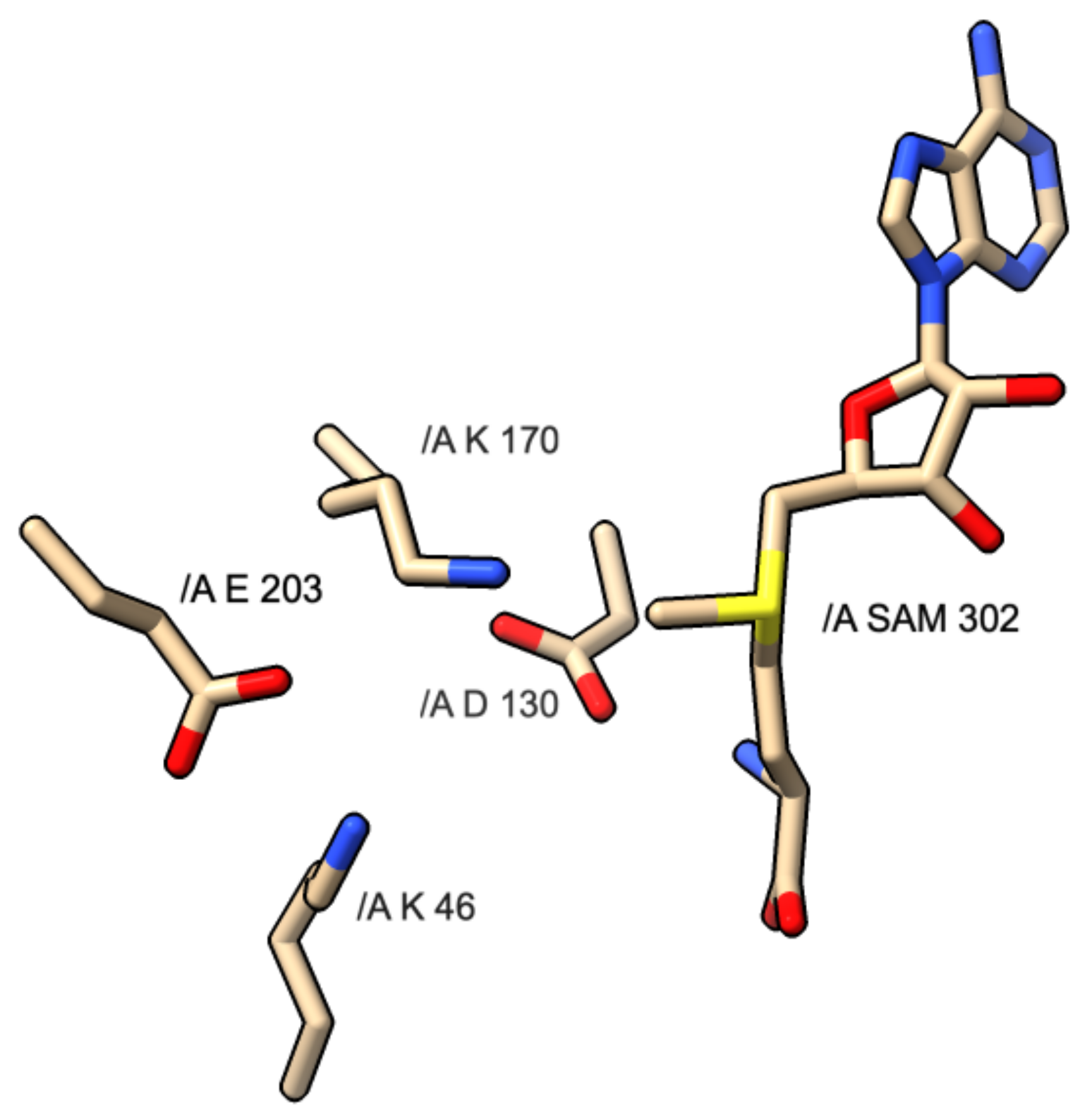
Figure 4.
Diagram of KDKE motif and interaction with SAM molecule found in PDB 3R24: SARS CoV-1 NSP16 protein.
Figure 4.
Diagram of KDKE motif and interaction with SAM molecule found in PDB 3R24: SARS CoV-1 NSP16 protein.
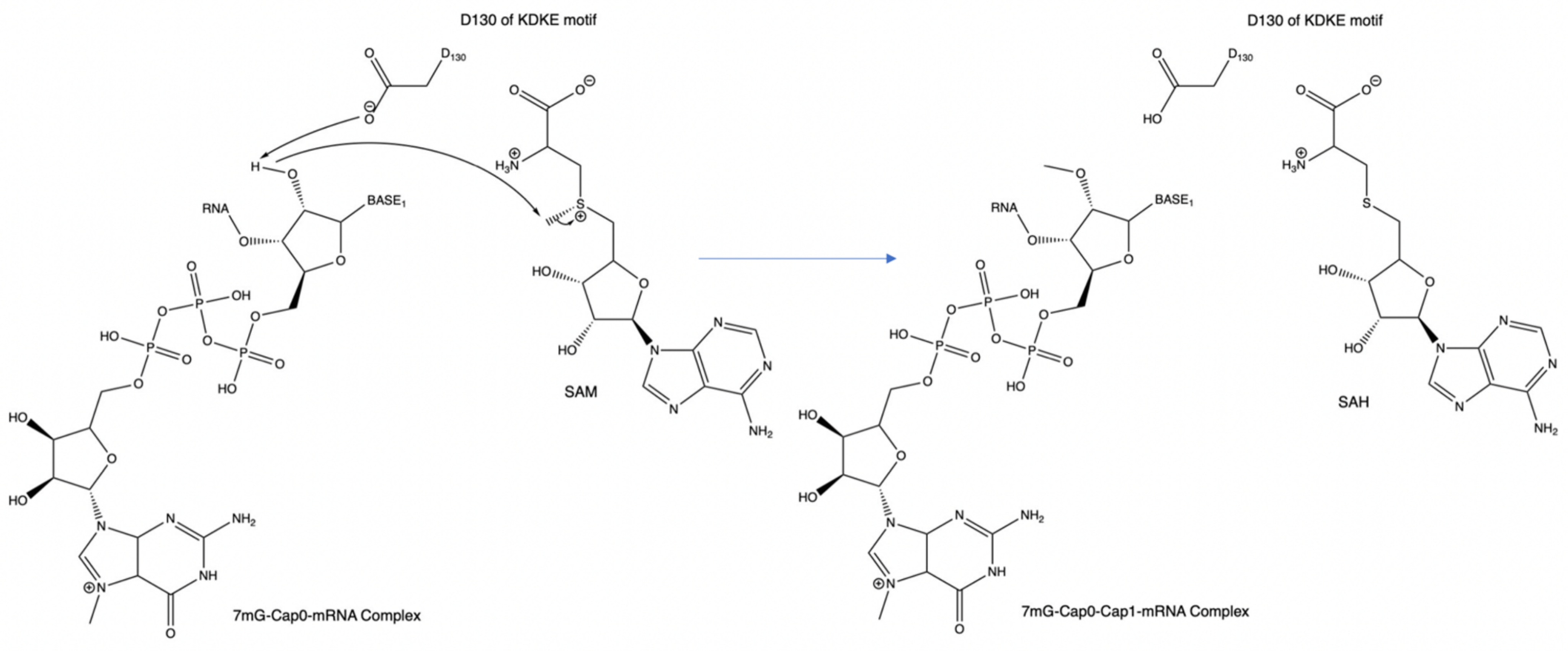
Figure 5.
An example of the mechanism of the KDKE motif within PDB 3R24. This can also be considered the general mechanism for methyl transfer onto a substrate.
Figure 5.
An example of the mechanism of the KDKE motif within PDB 3R24. This can also be considered the general mechanism for methyl transfer onto a substrate.
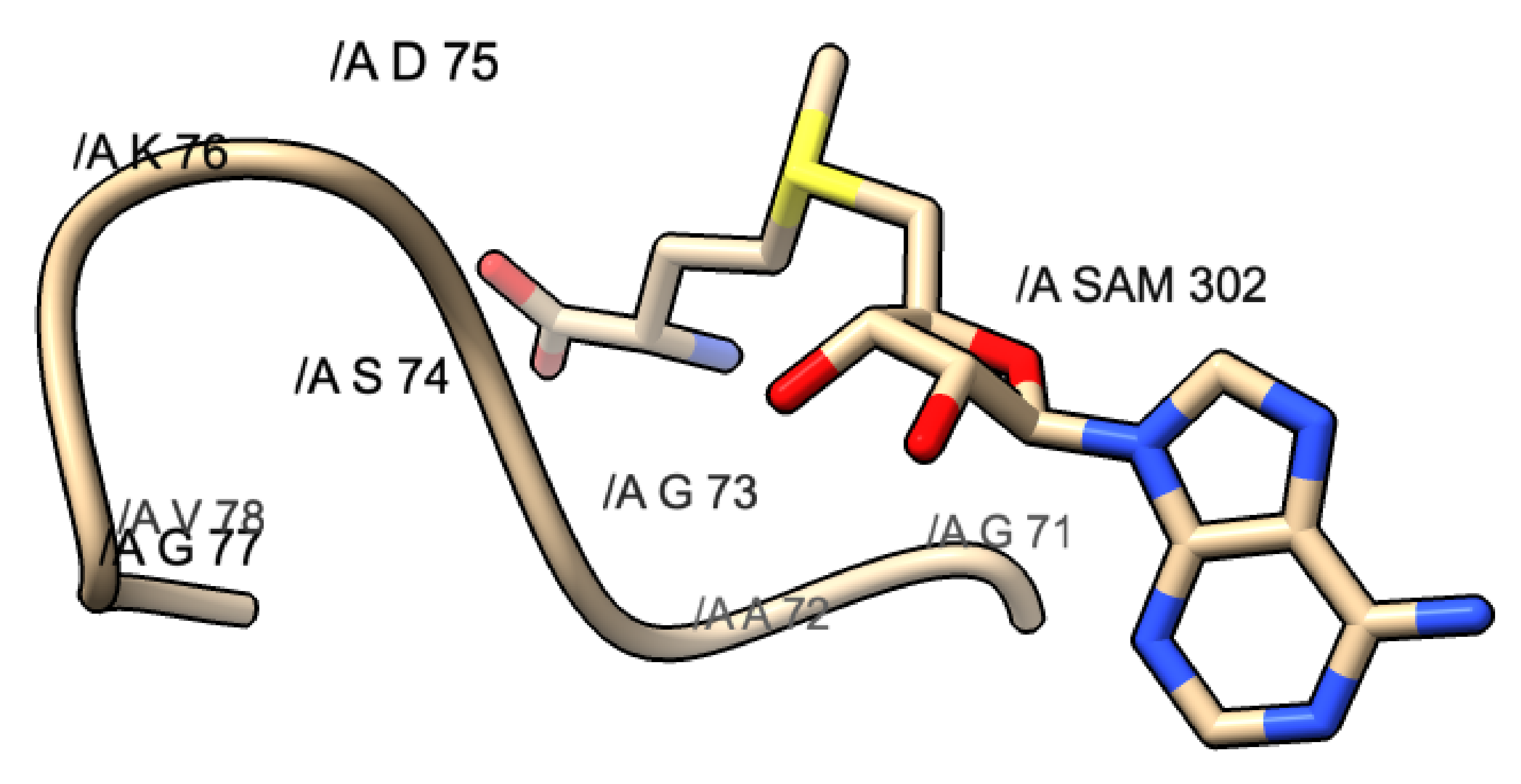
Figure 6.
GxG motif found in PDB 3R24: SARS-CoV. This motif is composed of G71, A72, and G73. The yellow shaded portion represents the methionine portion of SAM, the blue represents the nitrogen, and the red represents the oxygen atoms.
Figure 6.
GxG motif found in PDB 3R24: SARS-CoV. This motif is composed of G71, A72, and G73. The yellow shaded portion represents the methionine portion of SAM, the blue represents the nitrogen, and the red represents the oxygen atoms.
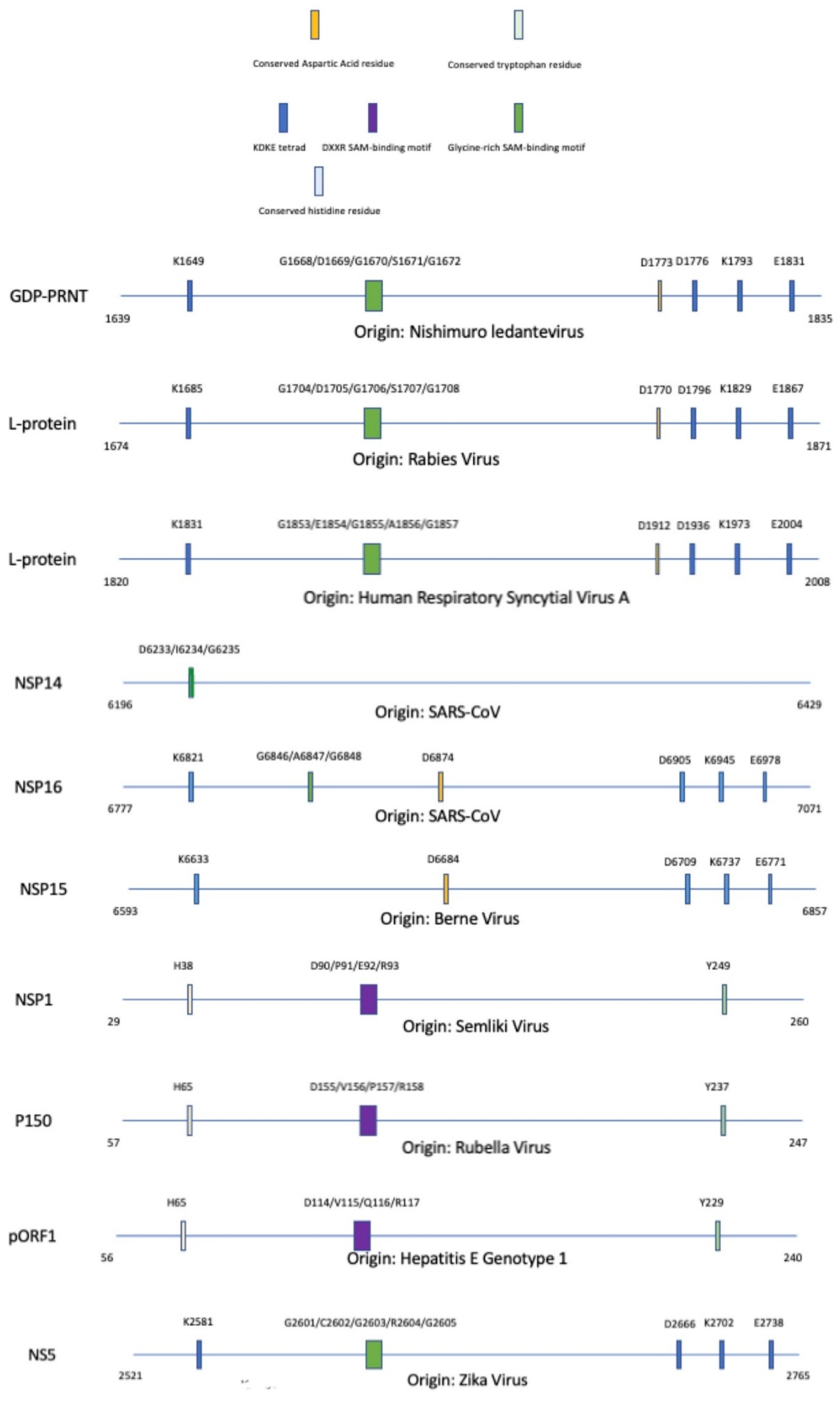
Figure 7.
Alignment of methyltransferase domains and their unique motifs involved with the completion of enzymatic activity; SAM: S-adenosyl-L-methionine. A legend is provided to partition each methyltransferase based on specific motifs that are conserved within the protein sequence. The significance of these motifs are explained throughout this paper.
Figure 7.
Alignment of methyltransferase domains and their unique motifs involved with the completion of enzymatic activity; SAM: S-adenosyl-L-methionine. A legend is provided to partition each methyltransferase based on specific motifs that are conserved within the protein sequence. The significance of these motifs are explained throughout this paper.
Figure 8.
A diagram of the simplified genome for the Zika Virus: C (Capsid), PR (peptide PR), M (small envelope protein), E (envelope protein), and NS protein (non-structural protein).
Figure 8.
A diagram of the simplified genome for the Zika Virus: C (Capsid), PR (peptide PR), M (small envelope protein), E (envelope protein), and NS protein (non-structural protein).
Figure 9.
A diagram of the simplified genome for the Semliki Virus: CP (Capsid protease), E (envelope protein), NS (non-structural protein), and 6 K protein (6 kDa protein).
Figure 9.
A diagram of the simplified genome for the Semliki Virus: CP (Capsid protease), E (envelope protein), NS (non-structural protein), and 6 K protein (6 kDa protein).
Figure 10.
A diagram of the simplified genome for the ORF1 polyprotein of Hepatitis E Virus: MT (methyltransferase), Y (Y-domain), PLP (papain-like protease), X (X-domain), Hel (Helicase), and RdRp (RNA-dependent RNA polymerase).
Figure 10.
A diagram of the simplified genome for the ORF1 polyprotein of Hepatitis E Virus: MT (methyltransferase), Y (Y-domain), PLP (papain-like protease), X (X-domain), Hel (Helicase), and RdRp (RNA-dependent RNA polymerase).
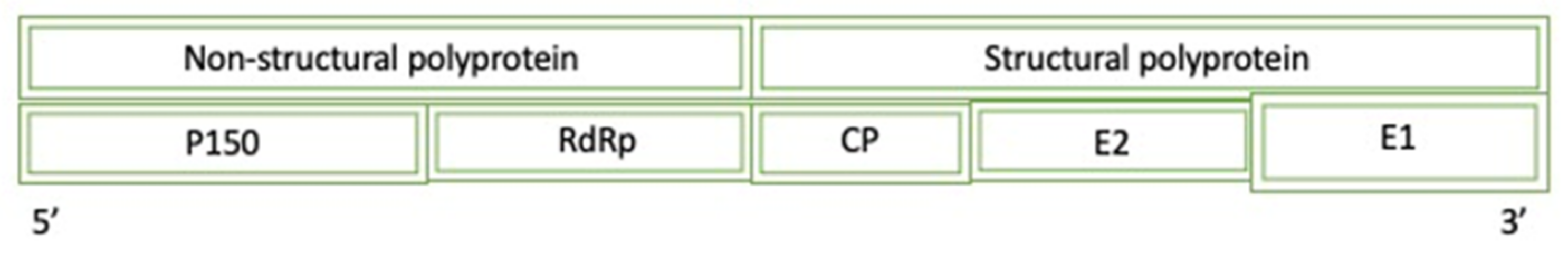
Figure 11.
A diagram of the simplified genome for the Rubella Virus: P150 (protease/methyltransferase 150), RdRp (RNA-dependent RNA polymerase), CP (cysteine protease), and E (envelope).
Figure 11.
A diagram of the simplified genome for the Rubella Virus: P150 (protease/methyltransferase 150), RdRp (RNA-dependent RNA polymerase), CP (cysteine protease), and E (envelope).
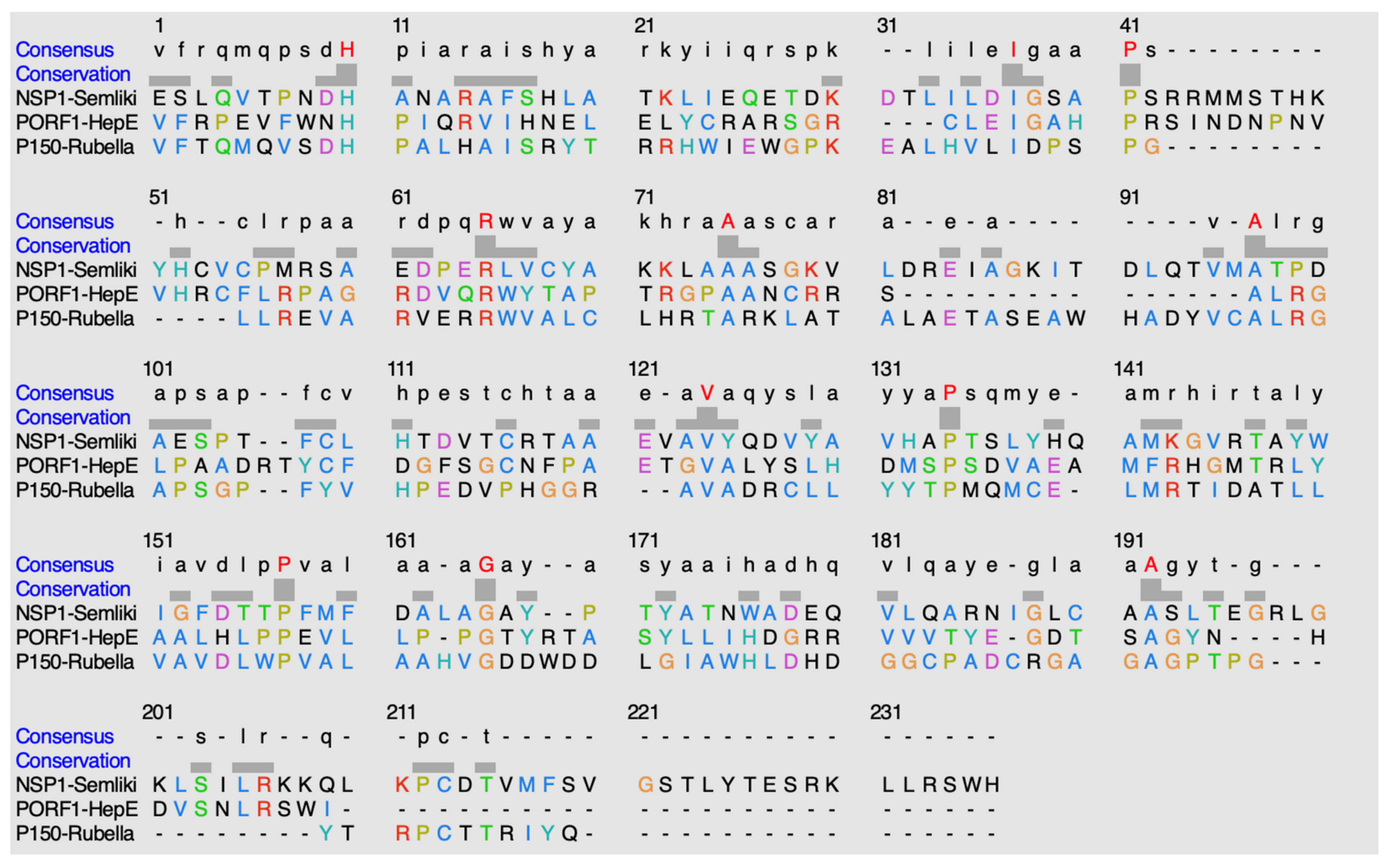
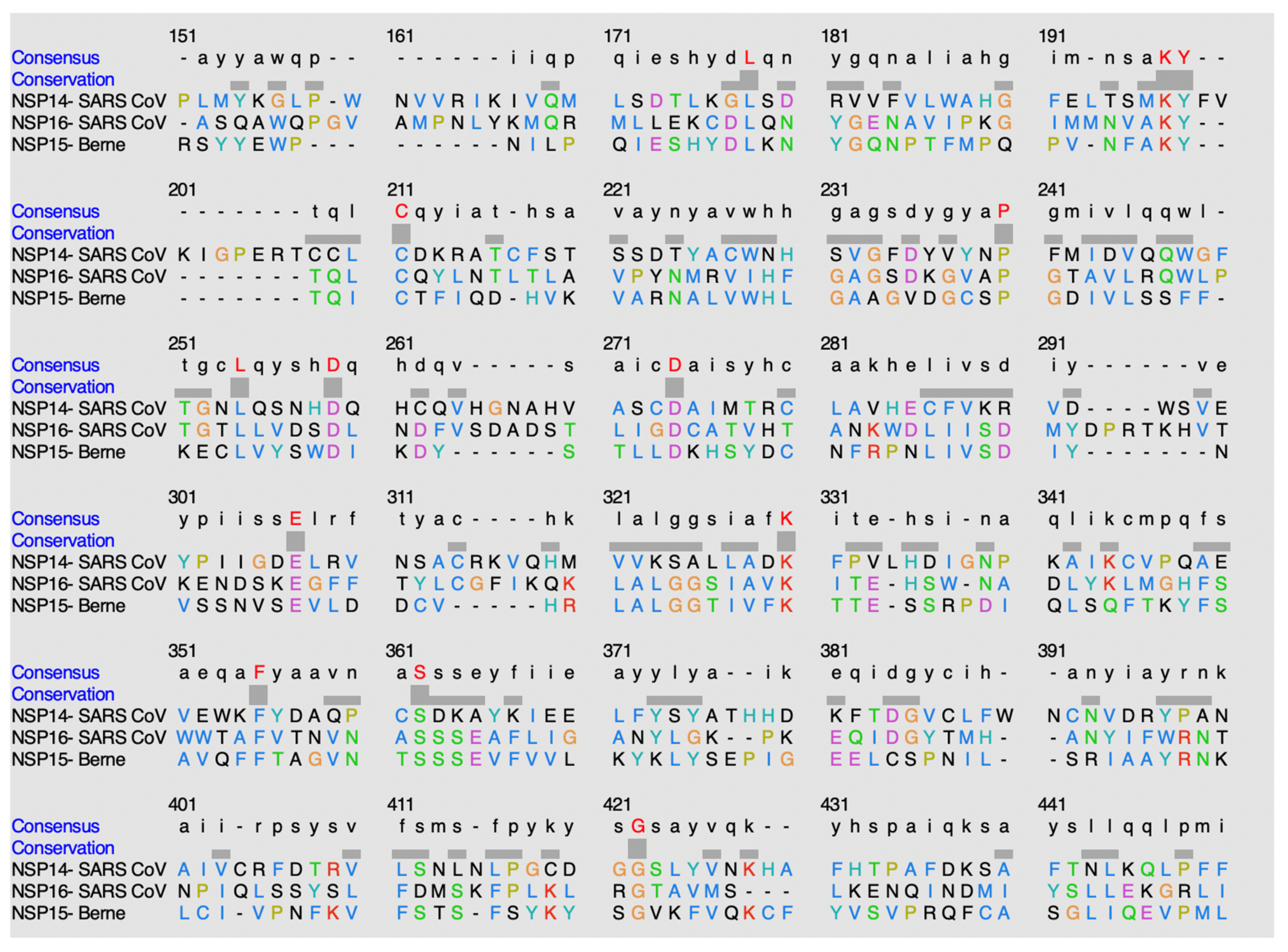
Figure 12.
Alignment of sequences using ClustalO. The highlighted red residues in the consensus row display residues that are highly conserved amongst the methyltransferases. The hyphens indicate either no consensus residue or no residue present at all.
Figure 12.
Alignment of sequences using ClustalO. The highlighted red residues in the consensus row display residues that are highly conserved amongst the methyltransferases. The hyphens indicate either no consensus residue or no residue present at all.
Figure 13.
A diagram of the simplified genome for the SARS-CoV Virus: NS (non-structural protein), PLpro (papain-like protease), 3CL (3CL-like protease), and RdRp (RNA-dependent RNA polymerase), Hel (Helicase).
Figure 13.
A diagram of the simplified genome for the SARS-CoV Virus: NS (non-structural protein), PLpro (papain-like protease), 3CL (3CL-like protease), and RdRp (RNA-dependent RNA polymerase), Hel (Helicase).
Figure 14.
A diagram of the simplified genome of ORF1ab for the Berne Virus: NS (non-structural protein). NSP refer to the non-structural proteins. ORF refers to the open-reading frame.
Figure 14.
A diagram of the simplified genome of ORF1ab for the Berne Virus: NS (non-structural protein). NSP refer to the non-structural proteins. ORF refers to the open-reading frame.
Figure 15.
Alignment of sequences using ClustalO.
Figure 15.
Alignment of sequences using ClustalO.
Figure 16.
A diagram of the simplified genome for the Human Respiratory Syncytial virus A: L (L protein), M2-1/2 (Protein M2-1), F (fusion glycoprotein), G (major surface glycoprotein), SH (small hydrophobic protein), M (matrix protein), P (phosphoprotein), N (nucleocapsid), NS (non-structural protein).
Figure 16.
A diagram of the simplified genome for the Human Respiratory Syncytial virus A: L (L protein), M2-1/2 (Protein M2-1), F (fusion glycoprotein), G (major surface glycoprotein), SH (small hydrophobic protein), M (matrix protein), P (phosphoprotein), N (nucleocapsid), NS (non-structural protein).
Figure 17.
A diagram of the simplified genome for the Nishimuro ledantevirus Virus: L (L protein), U1 (U1 protein), G (glycoprotein), M (matrix protein), P (phosphoprotein), (nucleocapsid).
Figure 17.
A diagram of the simplified genome for the Nishimuro ledantevirus Virus: L (L protein), U1 (U1 protein), G (glycoprotein), M (matrix protein), P (phosphoprotein), (nucleocapsid).
Figure 18.
A diagram of the simplified genome for the Rabies Virus: L (L protein), G (glycoprotein), M (matrix protein), P (phosphoprotein), N (nucleocapsid).
Figure 18.
A diagram of the simplified genome for the Rabies Virus: L (L protein), G (glycoprotein), M (matrix protein), P (phosphoprotein), N (nucleocapsid).
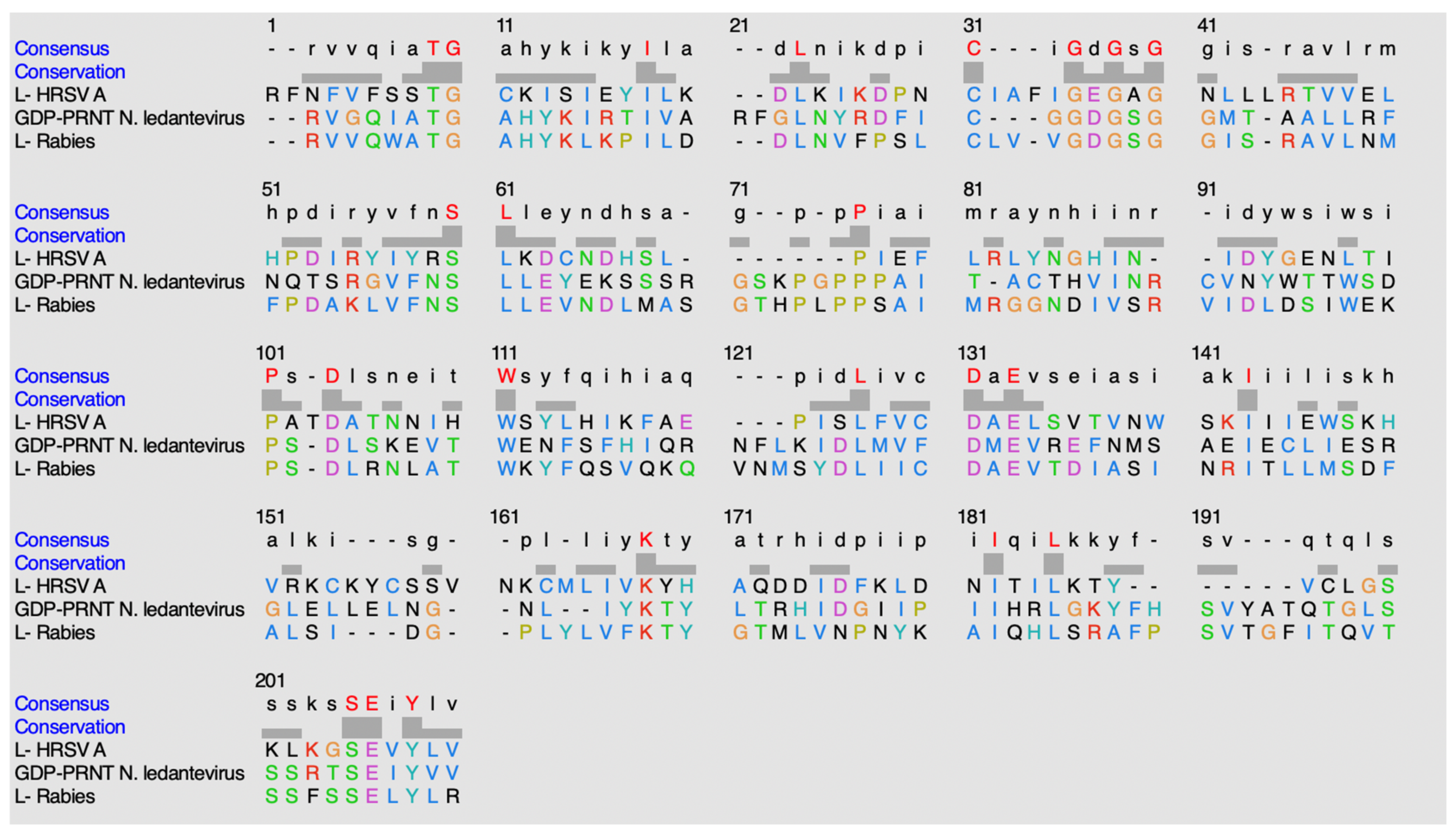
Figure 19.
Alignment sequence using ClustalO.
Figure 19.
Alignment sequence using ClustalO.
Figure 20.
Potential bi-substrate inhibitors that can inhibit both the SAM and mRNA binding pockets. (A) This image shows bi-substrate inhibitors that can act on NSP16 of SARS-CoV-2. (B) This image shows additional bi-substrate inhibitors that can act on NSP16 of SARS-CoV-2. (C) This image shows bi-substrate inhibitors that can act on NSP16 of SARS-CoV-2.
Figure 20.
Potential bi-substrate inhibitors that can inhibit both the SAM and mRNA binding pockets. (A) This image shows bi-substrate inhibitors that can act on NSP16 of SARS-CoV-2. (B) This image shows additional bi-substrate inhibitors that can act on NSP16 of SARS-CoV-2. (C) This image shows bi-substrate inhibitors that can act on NSP16 of SARS-CoV-2.
Figure 21.
This is a general scaffold design for methyltransferase inhibitors of SARS-CoV NSP16. Various R-groups have been tested through in silico screening and can be found in the paper referenced.
Figure 21.
This is a general scaffold design for methyltransferase inhibitors of SARS-CoV NSP16. Various R-groups have been tested through in silico screening and can be found in the paper referenced.
Figure 22.
The above inhibitors have the potential to inhibit DENV-3′s NS5 methyltransferase domain and have the potential to inhibit Zika’s NS5.
Figure 22.
The above inhibitors have the potential to inhibit DENV-3′s NS5 methyltransferase domain and have the potential to inhibit Zika’s NS5.
Figure 23.
Reported inhibitors of the Semliki NSP1 methyltransferase domain.
Figure 23.
Reported inhibitors of the Semliki NSP1 methyltransferase domain.
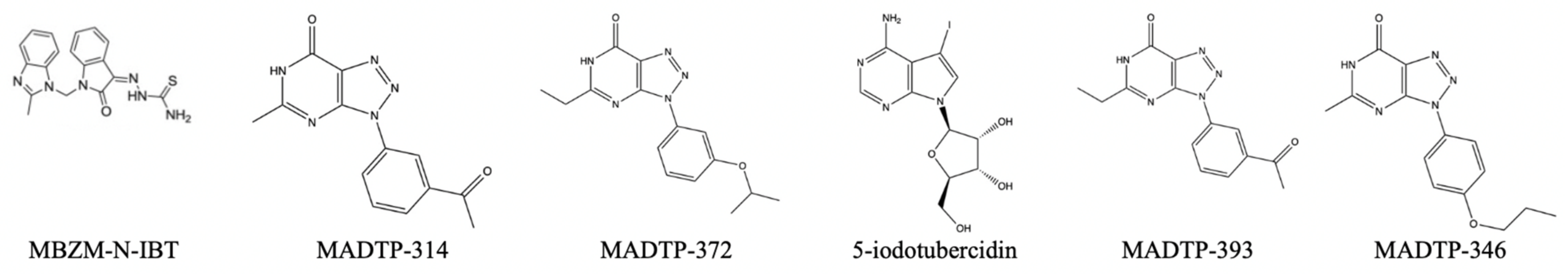
Figure 24.
Reported inhibitors of methyltransferases from the mononegavirales order [
31].
Figure 24.
Reported inhibitors of methyltransferases from the mononegavirales order [
31].
Table 1.
Summary of the Baltimore Classification System [
1].
Table 1.
Summary of the Baltimore Classification System [
1].
| Class | Genetic Material | Viral Examples |
|---|
| Class 1 | dsDNA | Poxviruses |
| Class 2 | ssDNA | Parvoviruses |
| Class 3 | dsRNA | Duplornaviricota |
| Class 4 | (+)-ssRNA | Kitrinoviricota |
| Class 5 | (–)-ssRNA | Orthornavirae |
| Class 6 | ssRNA-RT | Revtraviricetes |
| Class 7 | dsDNA-RT | Hepadnaviridae |
Table 2.
Summary of methyltransferase class type and their respective names [
6].
Table 2.
Summary of methyltransferase class type and their respective names [
6].
| Methyltransferase Structural Classes |
|---|
| Class Category | Name of Methyltransferase Family |
| Class 1 | Rossman-like alpha/beta |
| Class 2 | TIM beta/alpha-barrel alpha/beta |
| Class 3 | Tetrapyrrole methylase alpha/beta |
| Class 4 | SPOUT alpha/beta |
| Class 5 | SET domain all-beta |
| Class 6 | Transmembrane all alpha |
| Class 7 | DNA/RNA-binding 3-helical bundle all alpha |
| Class 8 | SSo0622-like alpha + beta |
| Class 9 | Thymidylate synthetase alpha + beta |
Table 3.
List of methyltransferases and corresponding viral example with Uniprot reference ID [
7].
Table 3.
List of methyltransferases and corresponding viral example with Uniprot reference ID [
7].
| | Characteristics of Single-Strand RNA Virus Methyltransferases |
|---|
| | Name of Methyltransferase | Viral Example | Uniprot ID |
|---|
| Positive-sense ssRNA | NSP14 | SARS-CoV | P0C6X7 |
| NSP16 | SARS-CoV | P0C6X7 |
| NSP15 | Berne Virus | P0C6V7 |
| NS5 | Zika Virus | A0A024B7W1 |
| P150 | Rubella Virus | Q86500 |
| NSP1 | Semliki Forest Virus | P08411 |
| pORF1 | Hepatitis E Genotype 1 | Q81862 |
| Negative-sense ssRNA | GDP-polyribonucleotidyltransferase | Nishimuro ledantevirus | R4X313 |
| L protein | Rabies virus | P16289 |
| L protein | Human Respiratory Syncytial Virus A | P28887 |
Table 4.
List of methyltransferases with the general role, function, substrate association, and assigned class [
7].
Table 4.
List of methyltransferases with the general role, function, substrate association, and assigned class [
7].
| Uniprot ID | Role | Reaction
Substrates | Purpose of Reaction | Class |
|---|
| P0C6X7 (NSP14) | This protein has the role of catalyzing the transfer of a methyl group from cofactor SAM to the N7 position of the first guanine nucleotide of mRNA. This methylation forms the cap-0 structure | SAM, lead guanine nucleotide | Using this methyltransferase allows for the formation of Cap-0 structure | Class 1-like SAM-binding methyltransferase superfamily |
| P0C6X7 (NSP16) | This protein has the role of catalyzing the transfer of a methyl group from cofactor SAM to 2′OH of the ribose following the initial guanine nucleotide of mRNA. This methylation forms the cap-1 structure | SAM, Cap-0 | Using this methyltransferase allows for the formation of Cap-1 structure | Class 1-like SAM-binding methyltransferase superfamily |
| P0C6V7 | This is a nidovirus-type SAM-dependent 2′-O-MTase | SAM, free ribose hydroxyl | This reaction forms the Cap-1 structure | Class 1-like SAM-binding methyltransferase superfamily |
| A0A024B7W1 | Catalyzes the methylation of guanine N-7 and ribose 2′-OH | SAM, GTP, cap-0 | Formation of Cap-0 and Cap-1 structures | mRNA cap 0–1 NS5-type methyltransferase family |
| Q86500 | This methyltransferase transfers a methyl group from SAM to viral mRNA | SAM, P150/P90 heterodimer, GTP | Formation of cap | Class 1-like SAM-binding methyltransferase superfamily |
| P08411 | Responsible for methylation of GMP at the C7 position | SAM, lead guanine nucleotide | nsP1 is responsible for methylation of the leading guanine nucleotide and then will complex itself to covalently link the cap to the mRNA | Class 1-like SAM-binding methyltransferase superfamily |
| Q81862 | This is an alpha-virus-like methyltransferase | SAM, GTP, GDP | Formation of cap | Class 1-like SAM-binding methyltransferase superfamily |
| R4X313 | This mononegavirus-type SAM-dependent 2′-O-MTase will methylate guanine-N7 and nucleoside-2-OH | SAM, GTP, ribose hydroxyl | Cap formation | Class 1-like SAM-binding methyltransferase superfamily |
| P16289 | This mononegavirus-type SAM-dependent 2′-O-MTase will methylate guanine-N7 and nucleoside-2-OH | SAM, GTP, Ribose-2′-OH | Cap formation | Class 1-like SAM-binding methyltransferase superfamily |
| P28887 | This multifunctional protein has both a ribose 2′-O methylation site and guanine-N7-methylation site. This will form both the Cap-0 and Cap-1 structures. Additionally, the enzyme is SAM-dependent | SAM, cap-0, lead guanine nucleotide | This reaction forms both the cap-0 and cap-1 structures | Class 1-like SAM-binding methyltransferase superfamily |
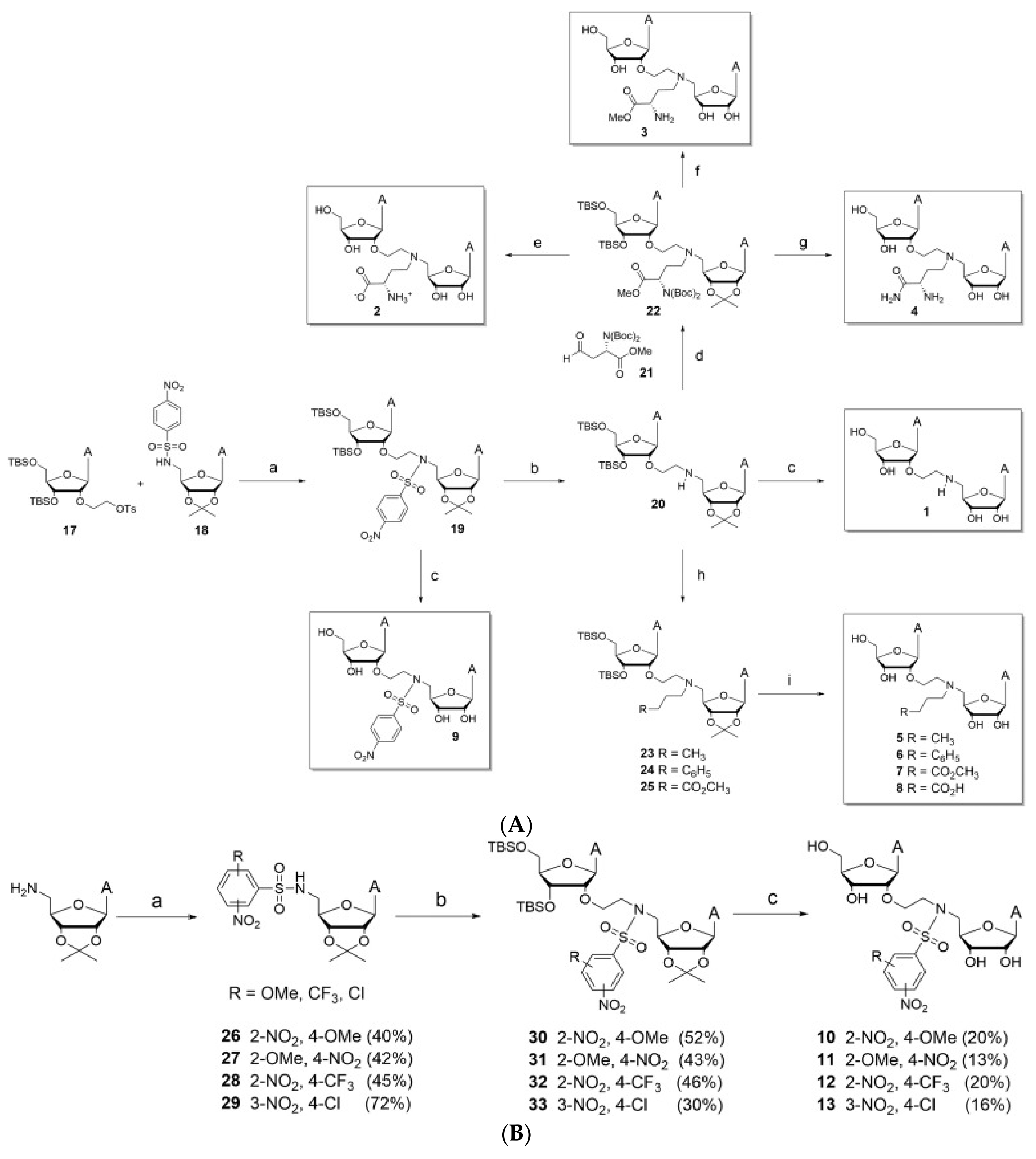
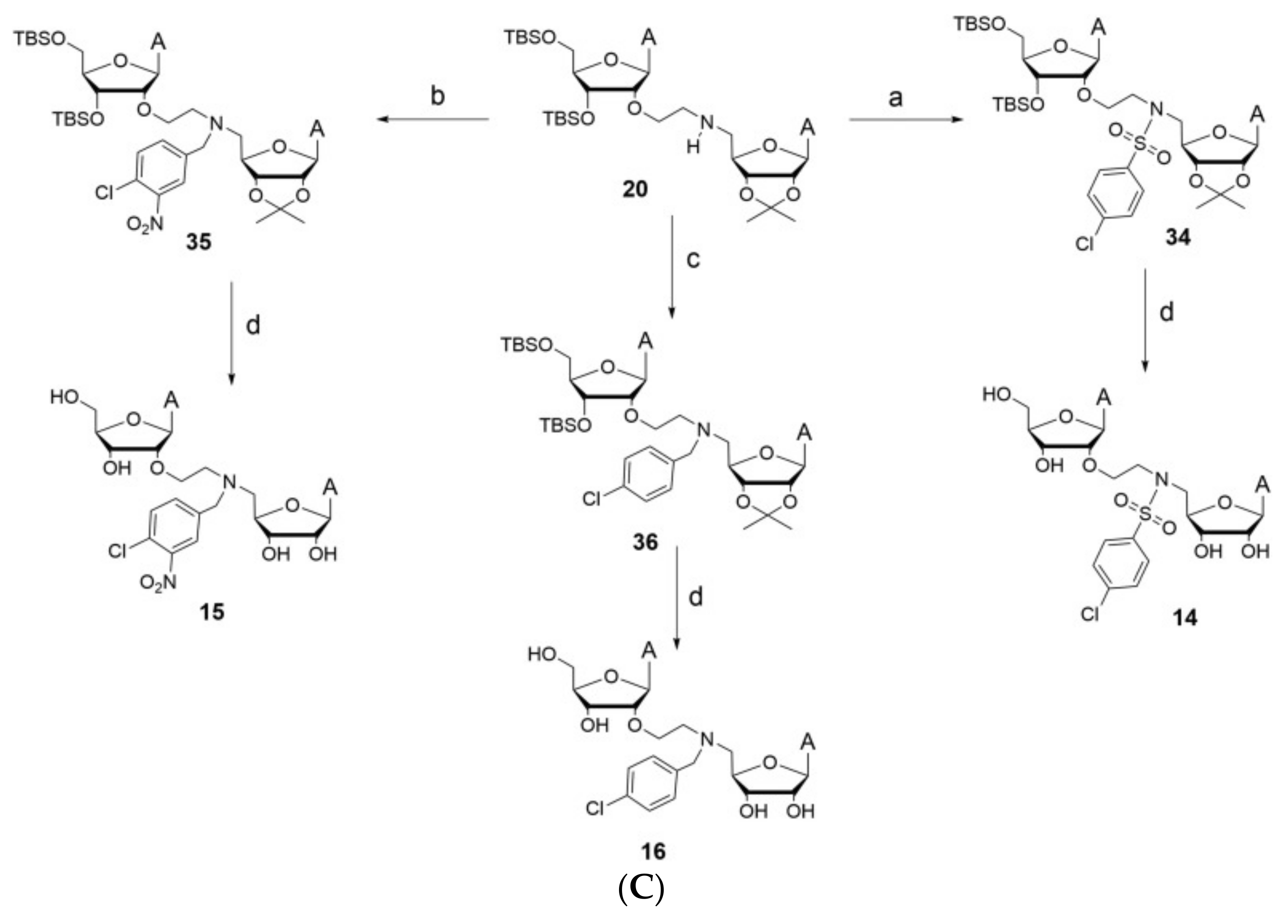
Table 5.
A table displaying the potential NSP14 inhibitor along with the measured percentage of inhibition of methyltransferase activity when using 50 um.
Table 5.
A table displaying the potential NSP14 inhibitor along with the measured percentage of inhibition of methyltransferase activity when using 50 um.
| Name of Molecule | Percentage of Inhibition at 50 um (%) |
|---|
| Sinefungin | 98.3 +/− 0.2 |
| 1 | 31.0 +/− 6.8 |
| 2 | 72.0 +/− 1.2 |
| 3 | 30.6 +/− 9.3 |
| 4 | 13.1 +/− 13.3 |
| 5 | No inhibition detected |
| 6 | 38.4 +/− 11.7 |
| 7 | No inhibition detected |
| 8 | 43.0 +/− 4.0 |
| 9 | 88.6 +/− 1.3 |
| 10 | 96.6 +/− 0.9 |
| 11 | 47.6 +/− 2.8 |
| 12 | 94.6 +/− 1.1 |
| 13 | 97.2 +/− 2.7 |
| 14 | 96.2 +/− 1.5 |
| 15 | 94.0 +/− 1.1 |
| 16 | 75.9 +/− 2.5 |
Table 6.
A table displaying the most active NSP14 inhibitors along with the measured IC50 value.
Table 6.
A table displaying the most active NSP14 inhibitors along with the measured IC50 value.
| Comparison of IC50 Values with the Most Active Reported Compounds |
|---|
| Name of molecule | IC50 (um) |
| Sinefungin | 0.36 |
| 2 | 40.6 +/− 3.2 |
| 6 | 55.5 +/− 5.1 |
| 9 | 2.6 +/− 0.2 |
| 10 | 3.9 +/− 0.4 |
| 11 | 70.4 +/− 4.9 |
| 12 | 5.7 +/− 0.5 |
| 13 | 0.6 +/− 0.1 |
| 14 | 1.5 +/− 0.1 |
| 15 | 2.4 +/− 0.2 |
| 16 | 9.9 +/− 0.9 |
Table 7.
QPlogS represents predicted aqueous solubility, QlogBB represents predicted log of the brain/blood partition coefficient, QPlogKp represents predicted skin permeability, and QPlogKhsa represents predicted binding to human serum albumin.
Table 7.
QPlogS represents predicted aqueous solubility, QlogBB represents predicted log of the brain/blood partition coefficient, QPlogKp represents predicted skin permeability, and QPlogKhsa represents predicted binding to human serum albumin.
| Compound (PubChemID) | QPlogS | QlogBB | QPlogKp | QPlogKhsa | Docking Score |
|---|
| 44367977 | 0.20 | −1.82 | −9.60 | −0.99 | −12.05 |
| 25203154 | 0.33 | −1.56 | −9.04 | −0.94 | −11.83 |
| 71008334 | 0.45 | −1.41 | −9.96 | −0.81 | −11.81 |
| 14728195 | 0.45 | −1.86 | −9.77 | −0.92 | −11.66 |
| 25200440 | 0.34 | −1.96 | −9.67 | −0.90 | −11.63 |
| 66856272 | 0.17 | −1.68 | −9.27 | −0.91 | −11.57 |
| 44601596 | −0.48 | −2.52 | −8.14 | −1.29 | −11.50 |
| 44601604 | −1.32 | −2.00 | −7.62 | −1.16 | −11.42 |
| 66855668 | 0.36 | −1.54 | −9.39 | −0.91 | −11.04 |
| 57126779 | −0.96 | −2.71 | −8.96 | −0.94 | −11.00 |
| 54016655 | −1.51 | −1.53 | −6.63 | −0.67 | −10.83 |
| 57324736 | −0.02 | −1.92 | −9.82 | −0.89 | −10.11 |
| 117805851 | −0.87 | −1.70 | −7.80 | −0.78 | −10.06 |
| 91397803 | −0.76 | −2.26 | −8.43 | −0.92 | −9.95 |
| 71444955 | −0.99 | −2.60 | −7.69 | −1.08 | −9.94 |
| Sinefungin | −0.39 | −3.17 | −11.50 | −1.29 | −8.09 |
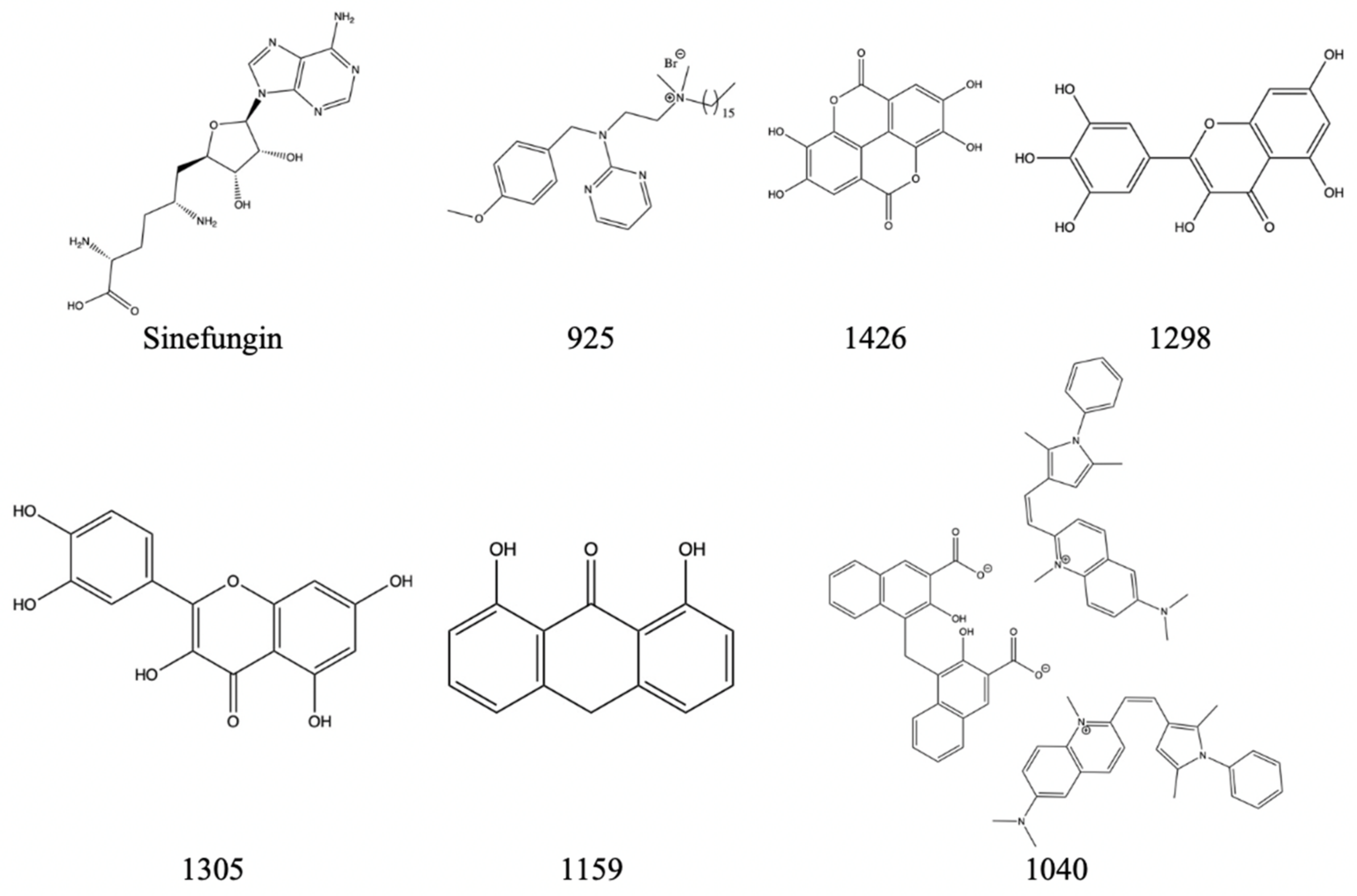
Table 8.
A table displaying the NS5 inhibitor compound name with its corresponding inhibition percentage at 50 um dose.
Table 8.
A table displaying the NS5 inhibitor compound name with its corresponding inhibition percentage at 50 um dose.
| Compound Name | Inhibition Percentage at 50 um (%) |
|---|
| Sinefungin | 98.3 |
| 925 | 98.9 |
| 1426 | 85 |
| 1298 | 44.9 |
| 1305 | 60.3 |
| 1159 | 37.6 |
| 1040 | 97.8 |
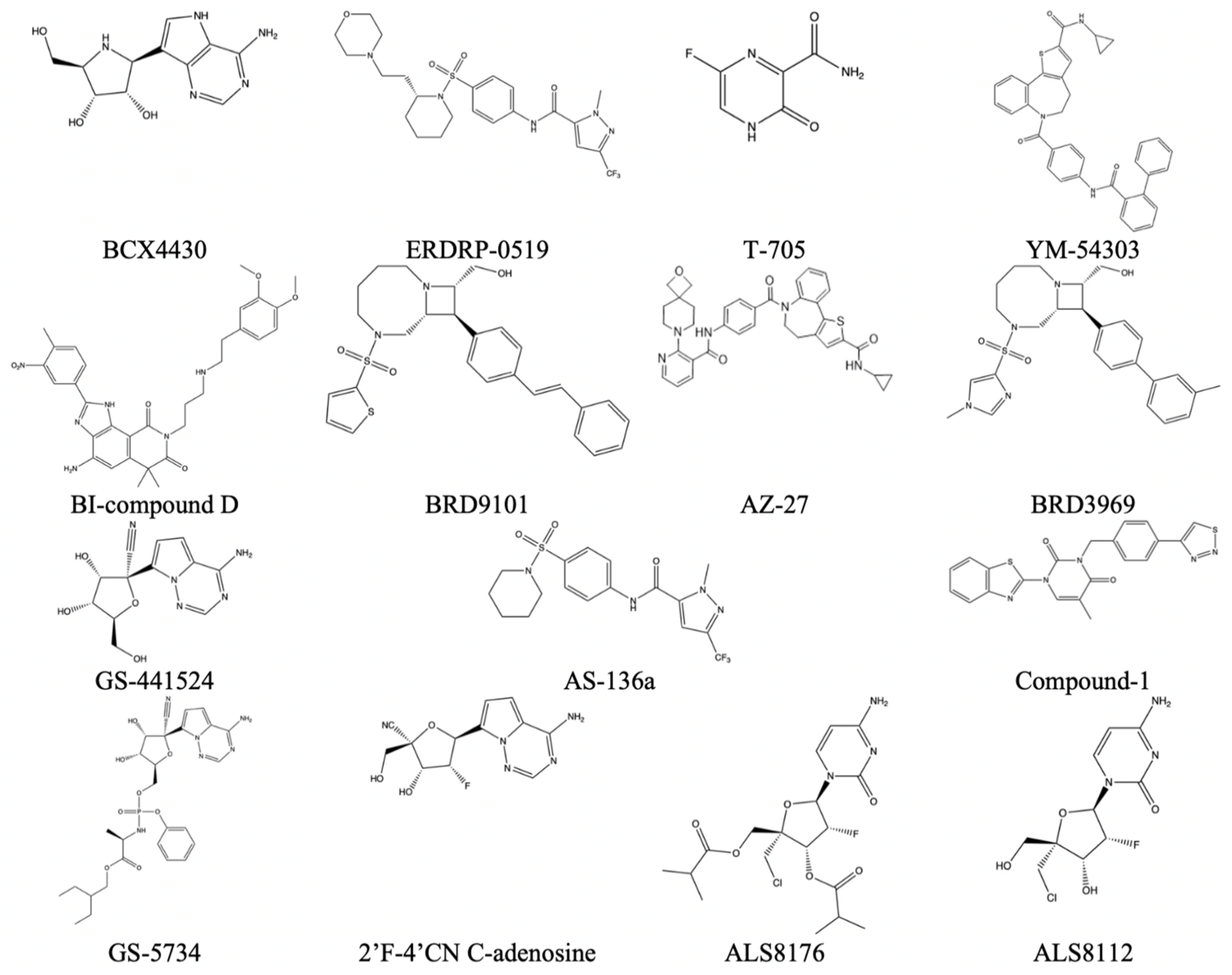
Table 9.
Reported virus, corresponding methyltransferase inhibitor, and EC50 value if available. This data is taken from Reference [
29].
Table 9.
Reported virus, corresponding methyltransferase inhibitor, and EC50 value if available. This data is taken from Reference [
29].
| Virus | Name of Molecule | EC50 (If Applicable) (µm) |
|---|
| HRSV-A | ALS-8112 | 0.15 µm |
| | ALS-8176 | 0.26 µm |
| | 2′F-4′CN C-adenosine | 2 µm |
| | BI-compound D | 0.021 µm |
| | YM-53403 | 0.72 µm |
| | AZ-27 | 0.01 µm |
| | Compound-1 | 1.6 µm |
| | BRD9101 | 1.7 µm |
| | BRD3969 | 1.6 µm |
| | T-705 | 260 µm |
| | BCX4430 | 11 µm |
| Measles | AS-136a | 0.014 µm |
| | ERDRP-0519 | 0.06 µm |
| Ebola | T-705 | 67 µm |
| | BCX4430 | 11.8 µm |
| | GS-441524 | 1.5 µm |
| | GS-5734 | 0.07 µm |





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}