
Antibodies Targeting KSHV gH/gL Reveal Distinct Neutralization Mechanisms


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Plasmids
2.3. Recombinant Proteins
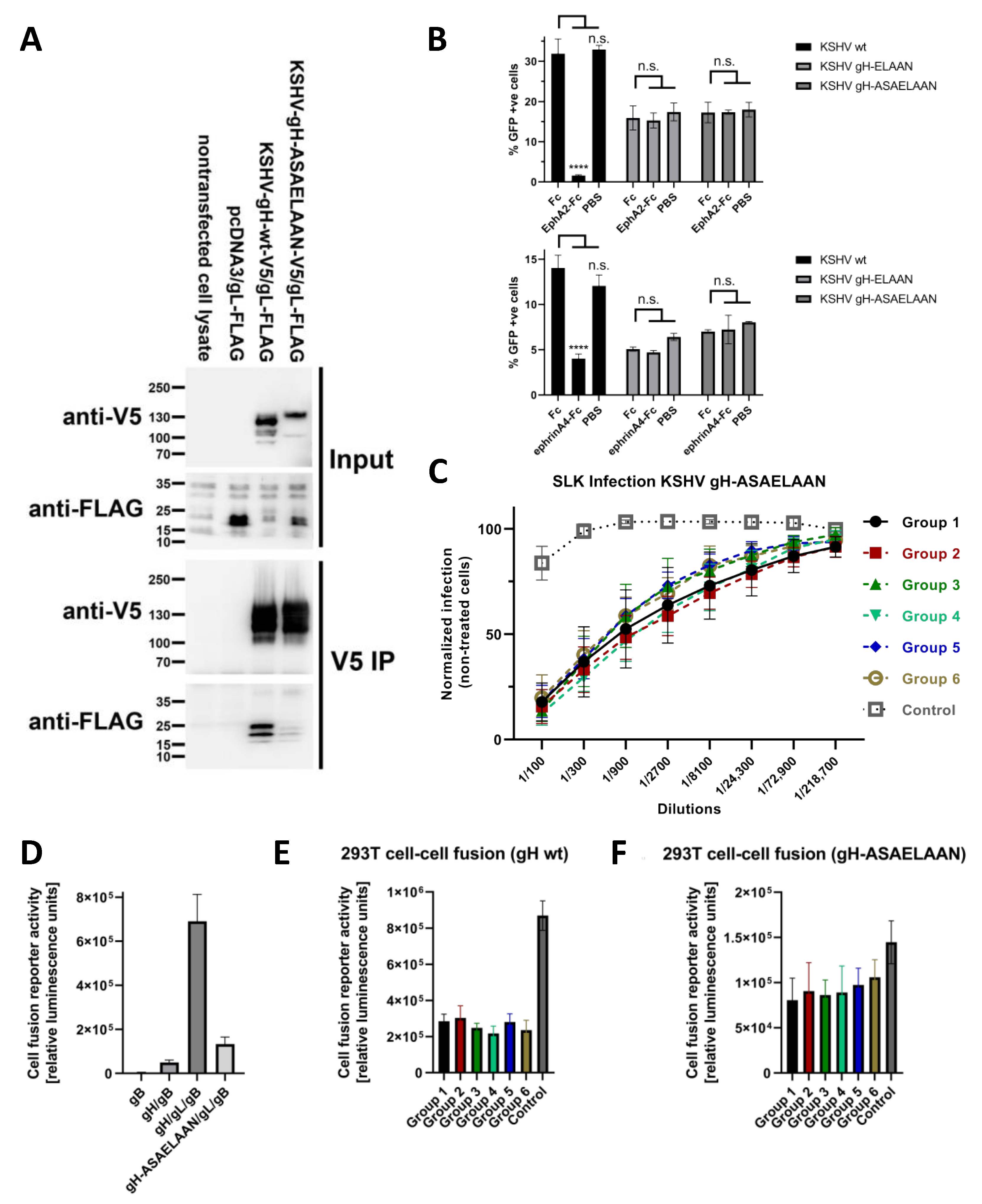
2.4. Immunoprecipitation, SDS Polyacrylamide Electrophoresis and Western Blot
2.5. Production of KSHV and KSHV gH-ASAELAAN
2.6. Immunization
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Fusion Assay
2.9. EphA2 Blocking Experiment
2.10. Infection Assays and Flow Cytometry
2.11. Statistical Analysis
3. Results
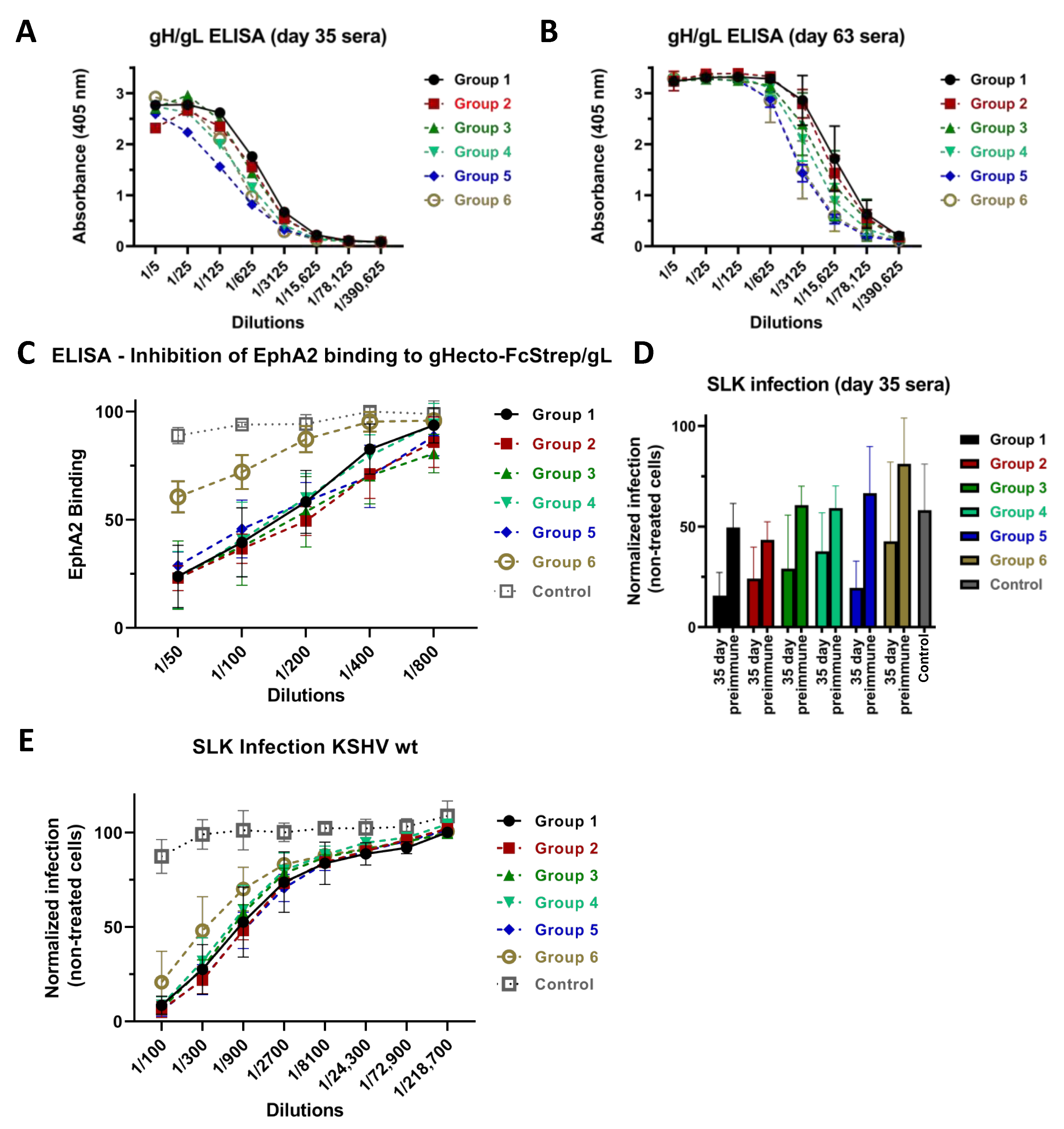
3.1. Immunization with Different Recombinant gH/gL Protein Complexes Elicits Binding Antibody Responses
3.2. Immunization with Recombinant gH/gL Elicits Antibodies That Block EphA2 Binding
3.3. Neutralizing Activity Is Induced by All Tested gH/gL Preparations
3.4. Antibodies That Do Not Block gH/gL Interactions with Eph Family Receptors or Target gL Contribute to Virus Neutralization
3.5. Sera Raised against gH/gL Inhibit Activation of gB for Fusion
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moore, P.S.; Chang, Y. Detection of Herpesvirus-like DNA Sequences in Kaposi’s Sarcoma in Patients with and without HIV Infection. N. Engl. J. Med. 1995, 332, 1181–1185. [Google Scholar] [CrossRef] [Green Version]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s Sarcoma and Its Associated Herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s Sarcoma-Associated Herpesvirus-like DNA Sequences in AIDS-Related Body-Cavity-Based Lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L. Kaposi’s Sarcoma-Associated Herpesvirus-like DNA Sequences in Multicentric Castleman’s Disease. Blood 1995, 86, 1276–1280. [Google Scholar] [CrossRef] [Green Version]
- Bower, M.; Nelson, M.; Young, A.M.; Thirlwell, C.; Newsom-Davis, T.; Mandalia, S.; Dhillon, T.; Holmes, P.; Gazzard, B.G.; Stebbing, J. Immune Reconstitution Inflammatory Syndrome Associated with Kaposi’s Sarcoma. J. Clin. Oncol. 2005, 23, 5224–5228. [Google Scholar] [CrossRef]
- Polizzotto, M.N.; Uldrick, T.S.; Hu, D.; Yarchoan, R. Clinical Manifestations of Kaposi Sarcoma Herpesvirus Lytic Activation: Multicentric Castleman Disease (KSHV-MCD) and the KSHV Inflammatory Cytokine Syndrome. Front. Microbiol. 2012, 3, 73. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Chen, J.; Li, Y.; Liu, D.; Zeng, Y.; Tian, Z.; Yunus, A.; Yang, Y.; Lu, J.; Song, X.; et al. Kaposi’s Sarcoma Herpesvirus Is Associated with Osteosarcoma in Xinjiang Populations. Proc. Natl. Acad. Sci. USA 2021, 118, e2016653118. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Nalwoga, A.; Webb, E.L.; Muserere, C.; Chihota, B.; Miley, W.; Labo, N.; Elliott, A.; Cose, S.; Whitby, D.; Newton, R. Variation in KSHV Prevalence between Geographically Proximate Locations in Uganda. Infect. Agents Cancer 2020, 15, 49. [Google Scholar] [CrossRef]
- Amir, H.; Kaaya, E.E.; Manji, K.P.; Kwesigabo, G.; Biberfeld, P. Kaposi’s Sarcoma before and during a Human Immunodeficiency Virus Epidemic in Tanzanian Children. Pediatr. Infect. Dis. J. 2001, 20, 518–521. [Google Scholar] [CrossRef]
- Ziegler, J.L.; Katongole-Mbidde, E. Kaposi’s Sarcoma in Childhood: An Analysis of 100 Cases from Uganda and Relationship to HIV Infection. Int. J. Cancer 1996, 65, 200–203. [Google Scholar] [CrossRef]
- Host, K.M.; Horner, M.-J.; van der Gronde, T.; Moses, A.; Phiri, S.; Dittmer, D.P.; Damania, B.; Gopal, S. Kaposi’s Sarcoma in Malawi: A Continued Problem for HIV-Positive and HIV-Negative Individuals. AIDS 2017, 31, 318–319. [Google Scholar] [CrossRef] [Green Version]
- Nalwoga, A.; Cose, S.; Wakeham, K.; Miley, W.; Ndibazza, J.; Drakeley, C.; Elliott, A.; Whitby, D.; Newton, R. Association between Malaria Exposure and Kaposi’s Sarcoma-Associated Herpes Virus Seropositivity in Uganda. Trop. Med. Int. Health 2015, 20, 665–672. [Google Scholar] [CrossRef]
- Blumenthal, M.J.; Schutz, C.; Meintjes, G.; Mohamed, Z.; Mendelson, M.; Ambler, J.M.; Whitby, D.; Mackelprang, R.D.; Carse, S.; Katz, A.A.; et al. EPHA2 Sequence Variants Are Associated with Susceptibility to Kaposi’s Sarcoma-Associated Herpesvirus Infection and Kaposi’s Sarcoma Prevalence in HIV-Infected Patients. Cancer Epidemiol. 2018, 56, 133–139. [Google Scholar] [CrossRef]
- Goedert, J.J.; Martin, M.P.; Vitale, F.; Lauria, C.; Whitby, D.; Qi, Y.; Gao, X.; Carrington, M. Risk of Classic Kaposi Sarcoma with Combinations of Killer Immunoglobulin-like Receptor and Human Leukocyte Antigen Loci: A Population-Based Case-Control Study. J. Infect. Dis. 2016, 213, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The Structural Basis of Herpesvirus Entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef]
- Hahn, A.; Birkmann, A.; Wies, E.; Dorer, D.; Mahr, K.; Stürzl, M.; Titgemeyer, F.; Neipel, F. Kaposi’s Sarcoma-Associated Herpesvirus GH/GL: Glycoprotein Export and Interaction with Cellular Receptors. J. Virol. 2009, 83, 396–407. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.S.; Kaufmann, J.K.; Wies, E.; Naschberger, E.; Panteleev-Ivlev, J.; Schmidt, K.; Holzer, A.; Schmidt, M.; Chen, J.; König, S.; et al. The Ephrin Receptor Tyrosine Kinase A2 Is a Cellular Receptor for Kaposi’s Sarcoma–Associated Herpesvirus. Nat. Med. 2012, 18, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.S.; Desrosiers, R.C. Rhesus Monkey Rhadinovirus Uses Eph Family Receptors for Entry into B Cells and Endothelial Cells but Not Fibroblasts. PLoS Pathog. 2013, 9, e1003360. [Google Scholar] [CrossRef]
- Großkopf, A.K.; Schlagowski, S.; Hörnich, B.F.; Fricke, T.; Desrosiers, R.C.; Hahn, A.S. EphA7 Functions as Receptor on BJAB Cells for Cell-to-Cell Transmission of the Kaposi’s Sarcoma-Associated Herpesvirus and for Cell-Free Infection by the Related Rhesus Monkey Rhadinovirus. J. Virol. 2019, 93, e00064-19. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, X.; Schaller, S.; Jardetzky, T.S.; Longnecker, R. Ephrin Receptor A4 Is a New Kaposi’s Sarcoma-Associated Herpesvirus Virus Entry Receptor. mBio 2019, 10, e02892-18. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.S.; Albrecht, J.C.; Birkmann, A.; Yağuboğlu, S.; Lang, D.; Fleckenstein, B.; Neipel, F. The Immunogenic Glycoprotein Gp35-37 of Human Herpesvirus 8 Is Encoded by Open Reading Frame K8.1. J. Virol. 1998, 72, 6725–6731. [Google Scholar] [CrossRef] [Green Version]
- Dollery, S.J.; Santiago-Crespo, R.J.; Chatterjee, D.; Berger, E.A. Glycoprotein K8.1A of Kaposi’s Sarcoma-Associated Herpesvirus Is a Critical B Cell Tropism Determinant, Independent of Its Heparan Sulfate Binding Activity. J. Virol. 2019, 93, e01876-18. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, Y.; Lidenge, S.J.; Tran, T.; West, J.T.; Wood, C.; Tso, F.Y. The Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) GH/GL Complex Is the Predominant Neutralizing Antigenic Determinant in KSHV-Infected Individuals. Viruses 2020, 12, 256. [Google Scholar] [CrossRef] [Green Version]
- Reeves, P.J.; Callewaert, N.; Contreras, R.; Khorana, H.G. Structure and Function in Rhodopsin: High-Level Expression of Rhodopsin with Restricted and Homogeneous N-Glycosylation by a Tetracycline-Inducible N-Acetylglucosaminyltransferase I-Negative HEK293S Stable Mammalian Cell Line. Proc. Natl. Acad. Sci. USA 2002, 99, 13419–13424. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, M.; Bu, W.; Joyce, M.G.; Meng, G.; Whittle, J.R.R.; Baxa, U.; Yamamoto, T.; Narpala, S.; Todd, J.-P.; Rao, S.S.; et al. Rational Design of an Epstein-Barr Virus Vaccine Targeting the Receptor-Binding Site. Cell 2015, 162, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Großkopf, A.K.; Ensser, A.; Neipel, F.; Jungnickl, D.; Schlagowski, S.; Desrosiers, R.C.; Hahn, A.S. A Conserved Eph Family Receptor-Binding Motif on the GH/GL Complex of Kaposi’s Sarcoma-Associated Herpesvirus and Rhesus Monkey Rhadinovirus. PLoS Pathog. 2018, 14, e1006912. [Google Scholar] [CrossRef] [Green Version]
- Hörnich, B.F.; Großkopf, A.K.; Dcosta, C.J.; Schlagowski, S.; Hahn, A.S. Interferon-Induced Transmembrane Proteins Inhibit Infection by the Kaposi’s Sarcoma-Associated Herpesvirus and the Related Rhesus Monkey Rhadinovirus in a Cell-Specific Manner. mBio 2021, 12, e0211321. [Google Scholar] [CrossRef]
- Großkopf, A.K.; Schlagowski, S.; Fricke, T.; Ensser, A.; Desrosiers, R.C.; Hahn, A.S. Plxdc Family Members Are Novel Receptors for the Rhesus Monkey Rhadinovirus (RRV). PLoS Pathog. 2021, 17, e1008979. [Google Scholar] [CrossRef]
- Longo, P.A.; Kavran, J.M.; Kim, M.-S.; Leahy, D.J. Transient Mammalian Cell Transfection with Polyethylenimine (PEI). Methods Enzymol. 2013, 529, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-Step Red-Mediated Recombination for Versatile High-Efficiency Markerless DNA Manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [PubMed]
- Brulois, K.F.; Chang, H.; Lee, A.S.-Y.; Ensser, A.; Wong, L.-Y.; Toth, Z.; Lee, S.H.; Lee, H.-R.; Myoung, J.; Ganem, D.; et al. Construction and Manipulation of a New Kaposi’s Sarcoma-Associated Herpesvirus Bacterial Artificial Chromosome Clone. J. Virol. 2012, 86, 9708–9720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensser, A.; Großkopf, A.K.; Mätz-Rensing, K.; Roos, C.; Hahn, A.S. Isolation and Sequence Analysis of a Novel Rhesus Macaque Foamy Virus Isolate with a Serotype-1-like Env. Arch. Virol. 2018, 163, 2507–2512. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Doria-Rose, N.A.; Cheng, C.; Stewart-Jones, G.B.E.; Chuang, G.-Y.; Chambers, M.; Druz, A.; Geng, H.; McKee, K.; Kwon, Y.D.; et al. Quantification of the Impact of the HIV-1-Glycan Shield on Antibody Elicitation. Cell Rep. 2017, 19, 719–732. [Google Scholar] [CrossRef]
- Cui, X.; Cao, Z.; Chen, Q.; Arjunaraja, S.; Snow, A.L.; Snapper, C.M. Rabbits Immunized with Epstein-Barr Virus GH/GL or GB Recombinant Proteins Elicit Higher Serum Virus Neutralizing Activity than Gp350. Vaccine 2016, 34, 4050–4055. [Google Scholar] [CrossRef]
- Bu, W.; Joyce, M.G.; Nguyen, H.; Banh, D.V.; Aguilar, F.; Tariq, Z.; Yap, M.L.; Tsujimura, Y.; Gillespie, R.A.; Tsybovsky, Y.; et al. Immunization with Components of the Viral Fusion Apparatus Elicits Antibodies That Neutralize Epstein-Barr Virus in B Cells and Epithelial Cells. Immunity 2019, 50, 1305–1316.e6. [Google Scholar] [CrossRef]
- Light, T.P.; Brun, D.; Guardado-Calvo, P.; Pederzoli, R.; Haouz, A.; Neipel, F.; Rey, F.A.; Hristova, K.; Backovic, M. Human Herpesvirus 8 Molecular Mimicry of Ephrin Ligands Facilitates Cell Entry and Triggers EphA2 Signaling. PLoS Biol. 2021, 19, e3001392. [Google Scholar] [CrossRef]
- Klupp, B.G.; Fuchs, W.; Weiland, E.; Mettenleiter, T.C. Pseudorabies Virus Glycoprotein L Is Necessary for Virus Infectivity but Dispensable for Virion Localization of Glycoprotein H. J. Virol. 1997, 71, 7687–7695. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Schaller, S.; Jardetzky, T.S.; Longnecker, R. EBV GH/GL and KSHV GH/GL Bind to Different Sites on EphA2 to Trigger Fusion. J. Virol. 2020, 94, e01454-20. [Google Scholar] [CrossRef]
- Hahn, A.S.; Bischof, G.F.; Großkopf, A.K.; Shin, Y.C.; Domingues, A.; Gonzalez-Nieto, L.; Rakasz, E.G.; Watkins, D.I.; Ensser, A.; Martins, M.A.; et al. A Recombinant Rhesus Monkey Rhadinovirus Deleted of Glycoprotein L Establishes Persistent Infection of Rhesus Macaques and Elicits Conventional T Cell Responses. J. Virol. 2020, 94, e01093-19. [Google Scholar] [CrossRef]
- Chang, P.-J.; Hung, C.-H.; Wang, S.-S.; Tsai, P.-H.; Shih, Y.-J.; Chen, L.-Y.; Huang, H.-Y.; Wei, L.-H.; Yen, J.-B.; Lin, C.-L.; et al. Identification and Characterization of Two Novel Spliced Genes Located in the Orf47-Orf46-Orf45 Gene Locus of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2014, 88, 10092–10109. [Google Scholar] [CrossRef] [PubMed] [Green Version]





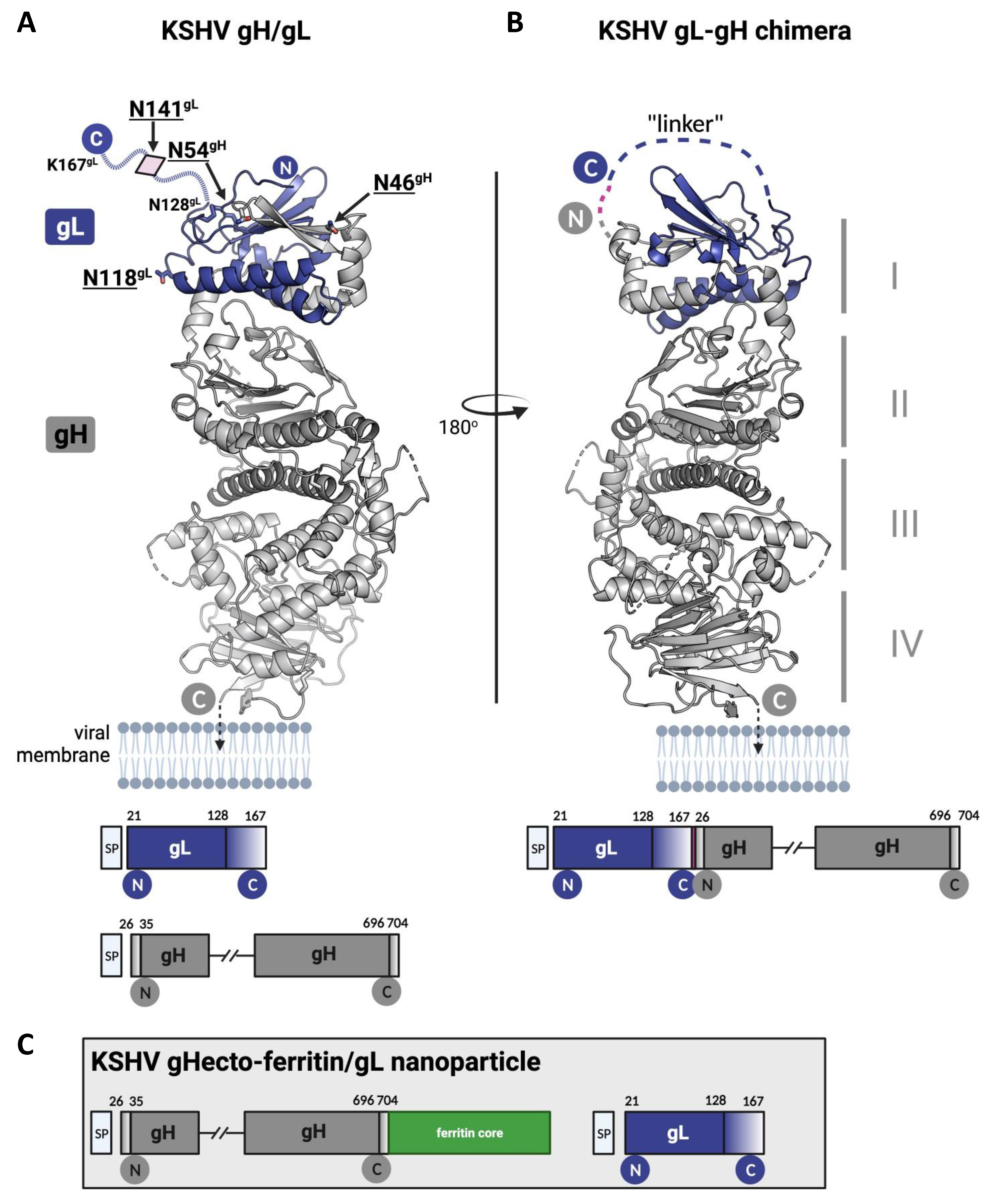{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotides | Sequence |
|---|---|
| gL N118Q for | AAGCCACCACAGCCGACAG |
| gL N118Q rev | GAAAGCCCACTGTATAGGCGGTC |
| gL N141Q for | AGCGGACCGGCTCTGTGAG |
| gL N141Q rev | GCATGGCCTTGCCCACAGAG |
| KSHVgH_N46Q_for | GCTGAGCATCGAGCTGGAATTC |
| KSHVgH_N46Q_rev | TGAGTCCGGCCGTTAATCAGC |
| KSHVgH_N54Q_for | GGGGACCTCCTTCTTTCTGAATTG |
| KSHVgH_N54Q_rev | TGGAATTCCAGCTCGATGCTCAG |
| for (gH-22) | AGCCCCGCAAGTCAGTGAG |
| rev (gH 23-) | ACTGGGGCTCTGCCTACC |
| for ([gH-22] gL 21-) | CTCACTGACTTGCGGGGCTTATGTCGCTCTGCCCTGTTGTG |
| rev (gL-167 [gH 23-]) | GTGGTAGGCAGAGCCCCAGTTTTCCCTTTCTGCCCTGCGTG |
| ferritin_for1KSHV | ACAGAAGGCGCGCCGCTTCTGAGAGTCAAGTCCGGCAAC |
| ferritin_rev | TTACCTTCGAAGGGCCCTTATCAGGACTTACGTGATTTCGC |
| KSHV_gH_for | AGAAGCGGCGCGCCTTCTG |
| gH_rev | TAAGGGCCCTTCGAAGGTAAGCC |
| Ax25-ASAELAANs | GCATCCGCTGAACTGGCAGCAAAC |
| Ax25-ASAELAANas | GTTGGTTCTCCCATTGATGAGCTGCG |
| EphA2-436 | CTGGTTGATGCTGACACTGGC |
| EphA2 rev2 | CATCATCACCATCACCATGAGTAAACC |
| Target | Details |
|---|---|
| V5-tag | Mouse, Bio-Rad, 1:1000, (secondary: Dianova, donkey anti-mouse 1:10,000) |
| DYKDDDDK (FLAG) tag | rabbit, Cell Signal Technology (D6W5B), 1:1000, (secondary: Dianova, goat anti-rabbit 1:10,000) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fricke, T.; Großkopf, A.K.; Ensser, A.; Backovic, M.; Hahn, A.S. Antibodies Targeting KSHV gH/gL Reveal Distinct Neutralization Mechanisms. Viruses 2022, 14, 541. https://doi.org/10.3390/v14030541
Fricke T, Großkopf AK, Ensser A, Backovic M, Hahn AS. Antibodies Targeting KSHV gH/gL Reveal Distinct Neutralization Mechanisms. Viruses. 2022; 14(3):541. https://doi.org/10.3390/v14030541
Chicago/Turabian StyleFricke, Thomas, Anna K. Großkopf, Armin Ensser, Marija Backovic, and Alexander S. Hahn. 2022. "Antibodies Targeting KSHV gH/gL Reveal Distinct Neutralization Mechanisms" Viruses 14, no. 3: 541. https://doi.org/10.3390/v14030541
APA StyleFricke, T., Großkopf, A. K., Ensser, A., Backovic, M., & Hahn, A. S. (2022). Antibodies Targeting KSHV gH/gL Reveal Distinct Neutralization Mechanisms. Viruses, 14(3), 541. https://doi.org/10.3390/v14030541