
Characterisation of Bacteriophage vB_SmaM_Ps15 Infective to Stenotrophomonas maltophilia Clinical Ocular Isolates

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Bacteriophage
2.3. Ps15 Titering and Plaque Morphology
2.4. S. maltophilia Bacterial Strain Fingerprinting
2.5. Host Range Analysis and the Efficiency of Plating (EOP)
2.6. Transmission Electron Microscopy (TEM)
2.7. Phage Assays
2.8. Phage Purification and DNA Isolation
2.9. Illumina MiSeq Phage Ps15 Sequencing and Genome Analysis
2.10. Comparative Genomics and Pangenome
2.11. Phylogenetics
3. Results
3.1. Ps15 Plaque Morphology
3.2. S. maltophilia Strain Fingerprinting
3.3. Host Range Analysis
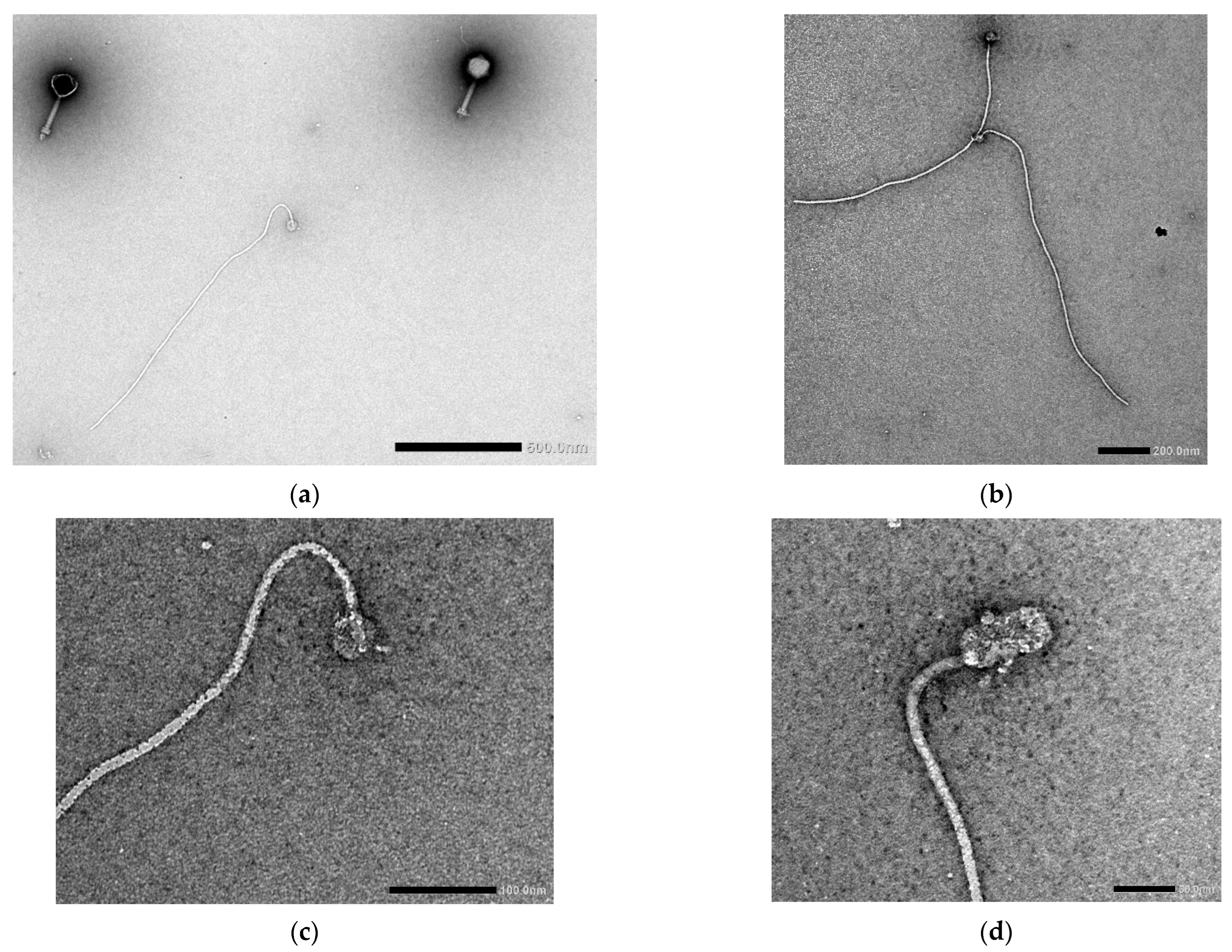
3.4. Transmission Electron Microscopy
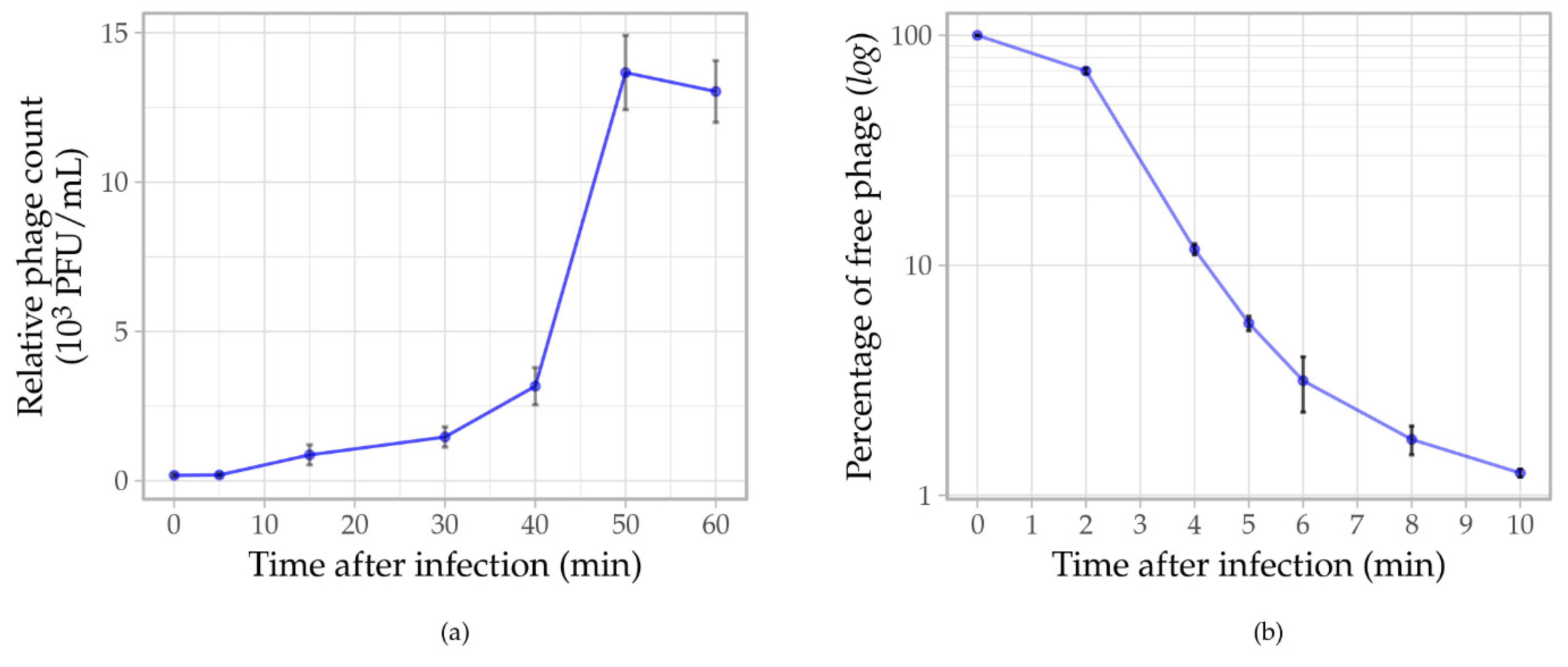
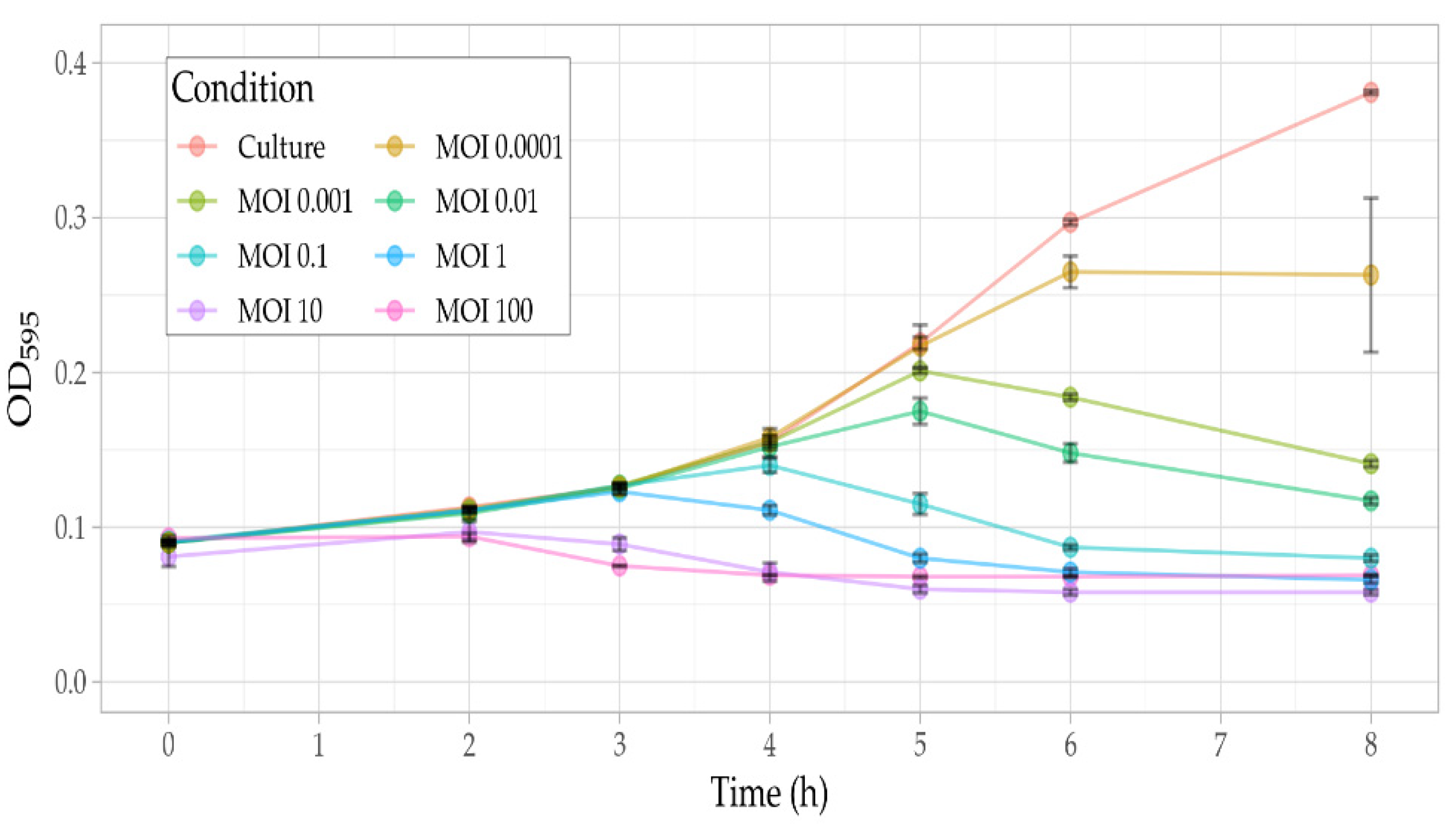
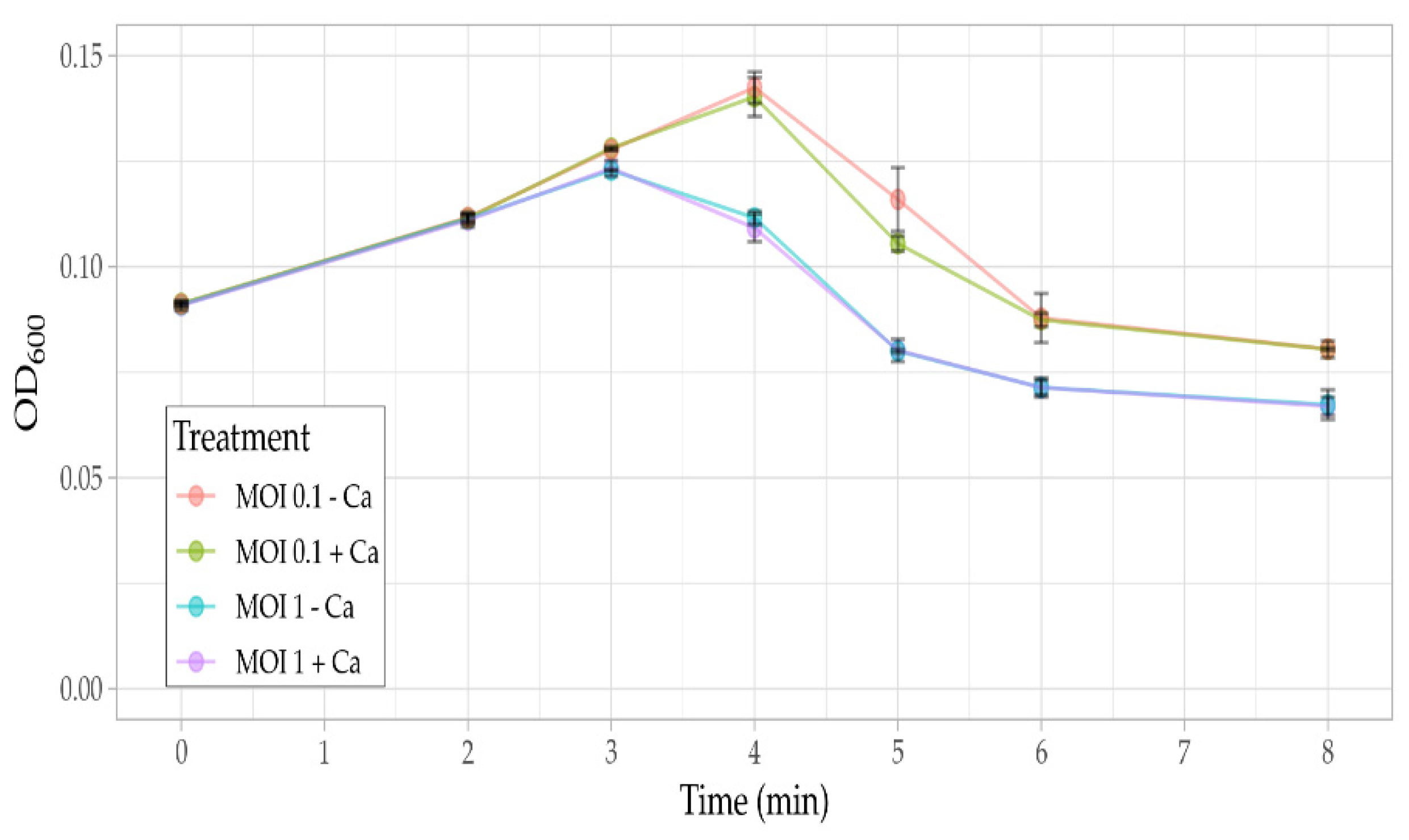
3.5. Phage Assays
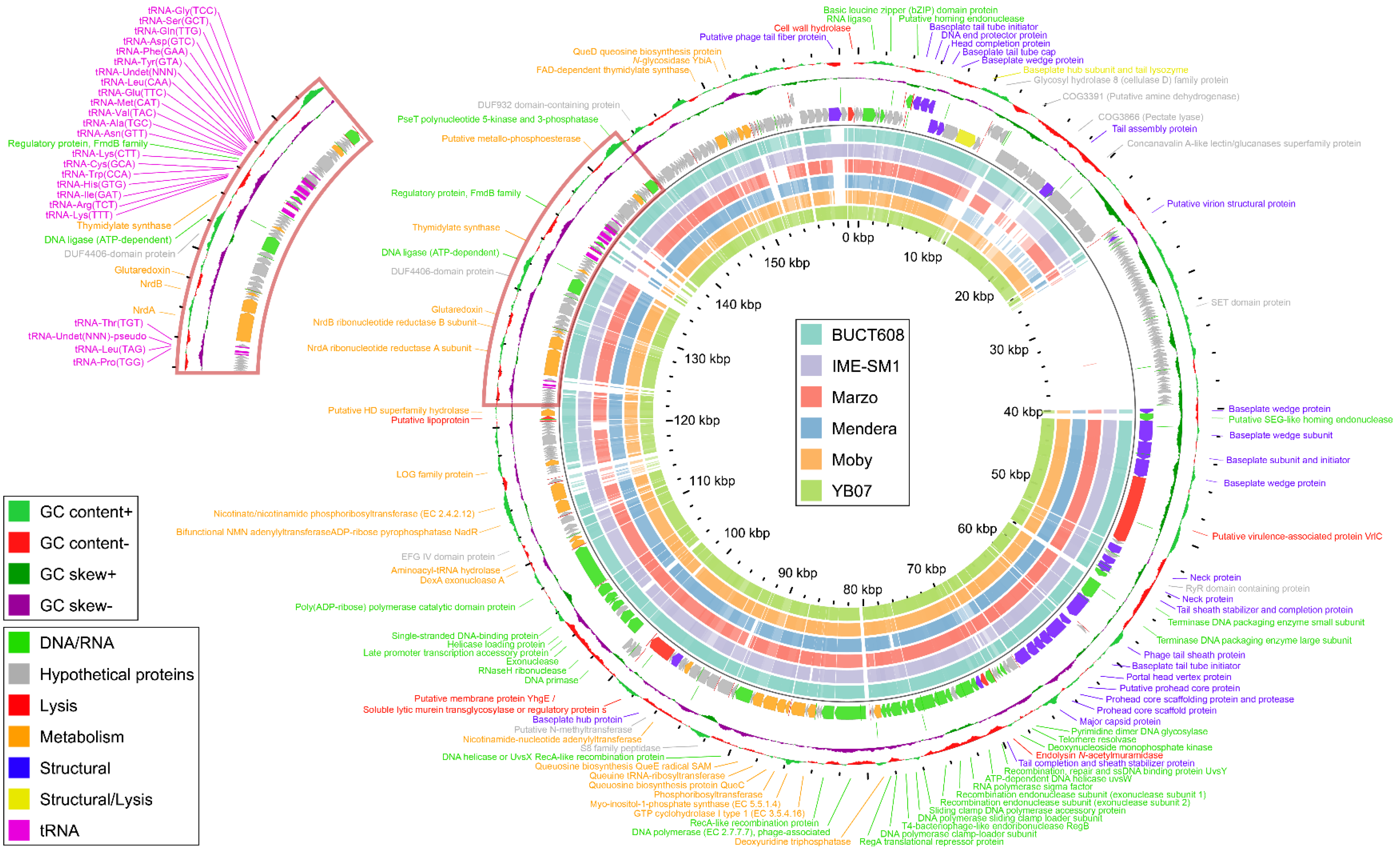
3.6. General Genome Characteristics
3.7. Functional Genome Analysis
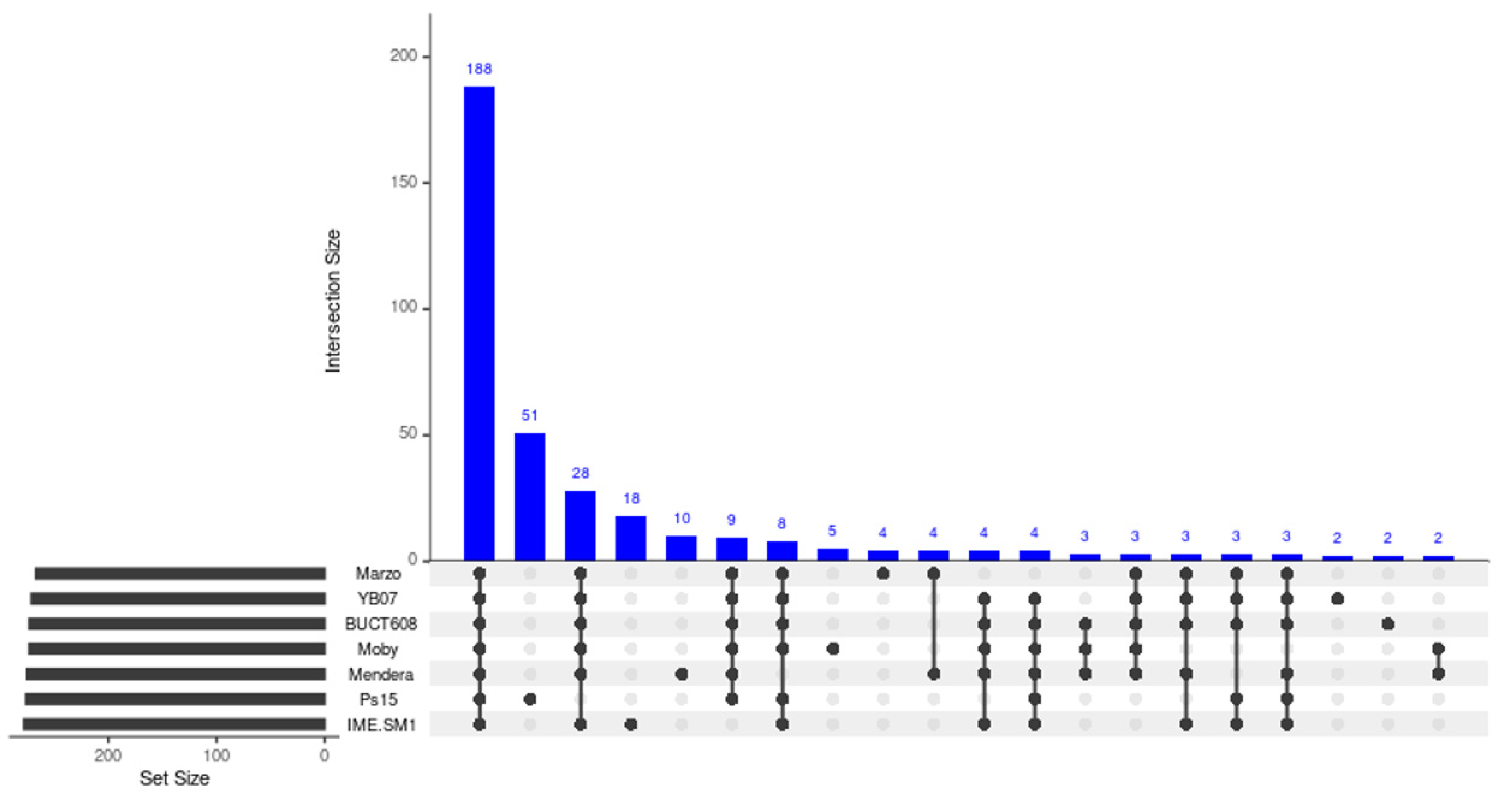
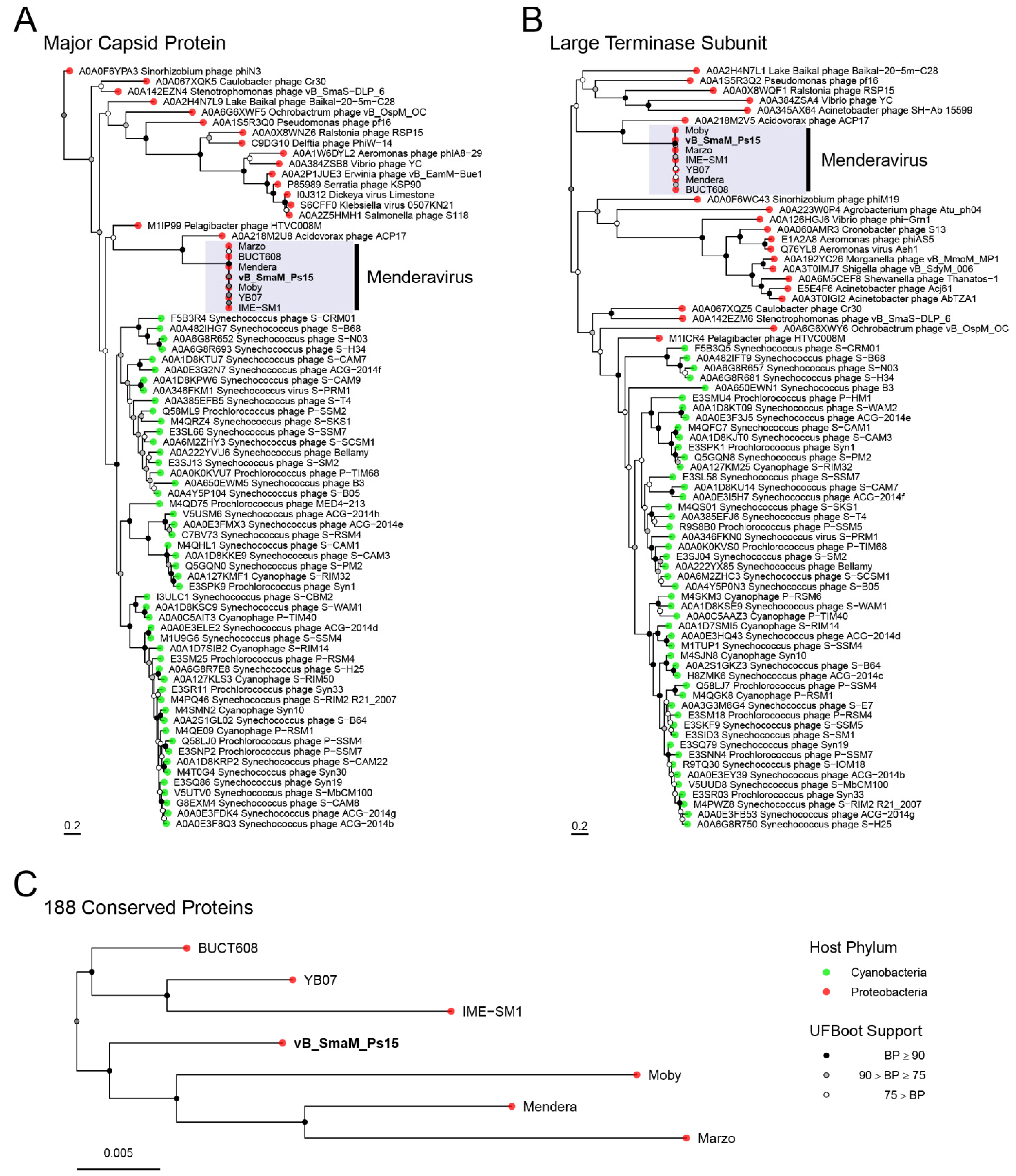
3.8. Comparative Genome Analysis and Phylogeny
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; van der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Brooke, J.S. Stenotrophomonas maltophilia: An emerging global opportunistic pathogen. Clin. Microbiol. Rev. 2012, 25, 2–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayol, A.; Corlouer, C.; Haenni, M.; Darty, M.; Maillard, K.; Desroches, M.; Lamy, B.; Jumas-Bilak, E.; Madec, J.Y.; Decousser, J.W. Are animals a source of Stenotrophomonas maltophilia in human infections? Contributions of a nationwide molecular study. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1039–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Prince, E.; Amin, W.F.; Thabet, S.S.; Hanna, M.I.L. Stenotrophomonas species in milk and some dairy products. J. Adv. Vet. Res. 2019, 9, 11–13. [Google Scholar]
- Adegoke, A.A.; Stenström, T.A.; Okoh, A.I. Stenotrophomonas maltophilia as an emerging ubiquitous pathogen: Looking beyond contemporary antibiotic therapy. Front. Microbiol. 2017, 30, 2276–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falagas, M.E.; Kastoris, A.C.; Vouloumanou, E.K.; Dimopoulos, G. Community-acquired Stenotrophomonas maltophilia infections: A systematic review. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 719–730. [Google Scholar] [CrossRef]
- Pathmanathan, A.; Waterer, G.W. Significance of positive Stenotrophomonas maltophilia culture in acute respiratory tract infection. Eur. Respir. J. 2005, 25, 911–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gales, A.C.; Jones, R.N.; Forward, K.R.; Liñares, J.; Sader, H.S.; Verhoef, J. Emerging importance of multidrug-resistant Acinetobacter species and Stenotrophomonas maltophilia as pathogens in seriously ill patients: Geographic patterns, epidemiological features, and trends in the SENTRY Antimicrobial Surveillance Program (1997–1999). Clin. Infect. Dis. 2001, 32, S104–S113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDaniel, M.S.; Schoeb, T.; Swords, W.E. Cooperativity between Stenotrophomonas maltophilia and Pseudomonas aeruginosa during polymicrobial airway infections. Infect. Immun. 2020, 88, e00855-19. [Google Scholar] [CrossRef] [PubMed]
- Rello, J.; Kalwaje Eshwara, V.; Lagunes, L.; Alves, J.; Wunderink, R.G.; Conway-Morris, A.; Rojas, J.N.; Alp, E.; Zhang, Z. A global priority list of the TOp TEn resistant microorganisms (TOTEM) study at intensive care: A prioritization exercise based on multi-criteria decision analysis. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Willcox, M.D. Management and treatment of contact lens-related Pseudomonas keratitis. Clin. Ophthalmol. 2012, 6, 919–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willcox, M.D.P.; Carnt, N.; Diec, J.; Naduvilath, T.; Evans, V.; Stapleton, F.; Iskandar, S.; Harmis, N.; Lazon de la Jara, P.; Holden, B.A. Contact lens case contamination during daily wear of silicone hydrogels. Optom. Vis. Sci. 2010, 87, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Vijay, A.K.; Willcox, M.D.P. Adhesion of Stenotrophomonas maltophilia, Delftia acidovorans and Achromobacter xylosoxidans to contact lenses. Eye Contact Lens 2018, 44, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Karaca, I.; Barut Selver, O.; Palamar, M.; Egrilmez, S.; Aydemir, S.; Yagci, A. Contact lens-associated microbial keratitis in a tertiary eye care center in Turkey. Eye Contact Lens 2020, 46, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.J.; Lim, H.R.; Koh, J.W. The status of infectious keratitis in Gwang-ju, Jeonnam, Republic of Korea. J. Korean Ophthalmol. Soc. 2021, 62, 173–183. [Google Scholar] [CrossRef]
- Cho, C.-H.; Lee, S.-B. Comparative analysis of the clinical aspects and treatment outcomes of Stenotrophomonas maltophilia keratitis and Pseudomonas aeruginosa keratitis. Eye Contact Lens 2021, 47, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Bottone, E.J.; Madayag, R.M.; Qureshi, M.N. Acanthamoeba keratitis: Synergy between amebic and bacterial cocontaminants in contact lens care systems as a prelude to infection. J. Clin. Microbiol. 1992, 30, 2447–2450. [Google Scholar] [CrossRef] [Green Version]
- Denet, E.; Vasselon, V.; Burdin, B.; Nazaret, S.; Favre-Bonté, S. Survival and growth of Stenotrophomonas maltophilia in free-living amoebae (FLA) and bacterial virulence properties. PLoS ONE 2018, 13, e0192308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagnier, I.; Raoult, D.; La Scola, B. Isolation and identification of amoeba-resisting bacteria from water in human environment by using an Acanthamoeba polyphaga co-culture procedure. Environ. Microbiol. 2008, 10, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, D.; Müller, K.-D.; Michel, R. Molecular characterization and ultrastructure of a new amoeba endoparasite belonging to the Stenotrophomonas maltophilia complex. Exp. Parasitol. 2013, 133, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Górski, A.; Miedzybrodzki, R.; Borysowski, J.; Weber-Dabrowska, B.; Lobocka, M.; Fortuna, W.; Letkiewicz, S.; Zimecki, M.; Filby, G. Bacteriophage therapy for the treatment of infections. Curr. Opin. Investig. Drugs 2009, 10, 766–774. [Google Scholar]
- Fukuda, K.; Ishida, W.; Uchiyama, J.; Rashel, M.; Kato, S.; Morita, T.; Muraoka, A.; Sumi, T.; Matsuzaki, S.; Daibata, M.; et al. Pseudomonas aeruginosa keratitis in mice: Effects of topical bacteriophage KPP12 administration. PLoS ONE 2012, 7, e47742. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, T.; Iwano, H.; Hiyashimizu, Y.; Matsubara, K.; Higuchi, H.; Nagahata, H.; Niwa, H.; Katayama, Y.; Kinoshita, Y.; Hagiwara, K.; et al. Phage therapy is effective in a mouse model of bacterial equine keratitis. Appl. Environ. Microbiol. 2016, 82, 5332–5339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, T.; Ishida, W.; Fukuda, K.; Nakajima, I.; Suzuki, T.; Uchiyama, J.; Matsuzaki, S.; Todokoro, D.; Daibata, M.; Fukushima, A. Therapeutic effects of intravitreously administered bacteriophage in a mouse model of endophthalmitis caused by vancomycin-sensitive or -resistant Enterococcus faecalis. Antimicrob. Agents Chemother. 2019, 63, e01088-19. [Google Scholar] [CrossRef] [PubMed]
- Urban-Chmiel, R.; Balicki, I.; Świąder, K.; Nowaczek, A.; Pyzik, E.; Stępień-Pyśniak, D.; Marek, A.; Puchalski, A.; Wernicki, A.; Poleszak, E.; et al. The in vitro efficacy of eye drops containing a bacteriophage solution specific for Staphylococcus spp. isolated from dogs with bacterial conjunctivitis. Ir. Vet. J. 2020, 73, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Fadlallah, A.; Chelala, E.; Legeais, J.M. Corneal infection therapy with topical bacteriophage administration. Open Ophthalmol. 2015, 9, 167–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCutcheon, J.G.; Dennis, J.J. The potential of phage therapy against the emerging opportunistic pathogen Stenotrophomonas maltophilia. Viruses 2021, 13, 1057. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Chen, C.R.; Lin, J.W.; Shen, G.H.; Chang, K.M.; Tseng, Y.H.; Weng, S.F. Isolation and characterization of novel giant Stenotrophomonas maltophilia phage phiSMA5. Appl. Environ. Microbiol. 2005, 71, 1387–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, D.; Karlene, L.; Stothard, P.; Dennis, J. The isolation and characterization of two Stenotrophomonas maltophilia bacteriophages capable of cross-taxonomic order infectivity. BMC Genom. 2015, 16, 664–678. [Google Scholar] [CrossRef] [Green Version]
- McCutcheon, J.G.; Lin, A.; Dennis, J.J. Isolation and characterization of the novel bacteriophage AXL3 against Stenotrophomonas maltophilia. Int. J. Mol. Sci. 2020, 21, 6338. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Zhu, H.; Willcox, M. Susceptibility of Stenotrophomonas maltophilia clinical isolates to antibiotics and contact lens multipurpose disinfecting solutions. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8475–8479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Stapleton, F.; Willcox, M.D.P. Susceptibility of contact lens-related Pseudomonas aeruginosa keratitis isolates to multipurpose disinfecting solutions, disinfectants, and antibiotics. Trans. Vis. Sci. Technol. 2020, 9, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Twest, R.; Kropinski, A.M. Bacteriophage enrichment from water and soil. Methods Mol. Biol. 2009, 501, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kropinski, A.M.; Prangishvili, D.; Lavigne, R. Position paper: The creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ. Microbiol. 2009, 11, 2775–2777. [Google Scholar] [CrossRef] [PubMed]
- Kropinski, A.M. Measurement of the rate of attachment of bacteriophage to cells. Methods Mol. Biol. 2009, 501, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Koeuth, T.; Versalovic, J.; Lupski, J.R. Differential subsequence conservation of interspersed repetitive Streptococcus pneumoniae BOX elements in diverse bacteria. Genome Res. 1995, 5, 408–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutter, E. Chapter 14: Phage host range and efficiency of plating. Methods Mol. Biol. 2009, 501, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Kropinski, A.M. Practical advice on the one-step growth curve. Methods Mol. Biol. 2018, 1681, 41–47. [Google Scholar] [CrossRef]
- Leskinen, K.; Tuomala, H.; Wicklund, A.; Horsma-Heikkinen, J.; Kuusela, P.; Skurnik, M.; Kiljunen, S. Characterization of vB_SauM-fRuSau02, a Twort-like bacteriophage isolated from a therapeutic phage cocktail. Viruses 2017, 9, 258. [Google Scholar] [CrossRef]
- Mahony, J.; Tremblay, D.M.; Labrie, S.J.; Moineau, S.; van Sinderen, D. Investigating the requirement for calcium during lactococcal phage infection. Int. J. Food Microbiol. 2015, 201, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Charlebois, P.; Gnerre, S.; Coole, M.G.; Lennon, N.J.; Levin, J.Z.; Qu, J.; Ryan, E.M.; Zody, M.C.; Henn, M.R. De novo assembly of highly diverse viral populations. BMC Genom. 2012, 13, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Ecale Zhou, C.L.; Malfatti, S.; Kimbrel, J.; Philipson, C.; McNair, K.; Hamilton, T.; Edwards, R.; Souza, B. multiPhATE: Bioinformatics pipeline for functional annotation of phage isolates. Bioinformatics 2019, 35, 4402–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Zhou, C.; Dinsdale, E.A.; Souza, B.; Edwards, R.A. PHANOTATE: A novel approach to gene identification in phage genomes. Bioinformatics 2019, 35, 4537–4542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [Green Version]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.-H.; McDermott, P.F.; et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef] [Green Version]
- de Jong, A.; Pietersma, H.; Cordes, M.; Kuipers, O.P.; Kok, J. PePPER: A webserver for prediction of prokaryote promoter elements and regulons. BMC Genom. 2012, 13, 299–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baerends, R.J.; Smits, W.K.; de Jong, A.; Hamoen, L.W.; Kok, J.; Kuipers, O.P. Genome2D: A visualization tool for the rapid analysis of bacterial transcriptome data. Genome Biol. 2004, 5, R37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingsford, C.L.; Ayanbule, K.; Salzberg, S.L. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007, 8, R22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockenberry, A.J.; Wilke, C.O. BACPHLIP: Predicting bacteriophage lifestyle from conserved protein domains. PeerJ 2021, 9, e11396. [Google Scholar] [CrossRef]
- Petkau, A.; Stuart-Edwards, M.; Stothard, P.; Van Domselaar, G. Interactive microbial genome visualization with GView. Bioinformatics 2010, 26, 3125–3126. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.D.; Tomii, K.; Katoh, K. Application of the MAFFT sequence alignment program to large data-reexamination of the usefulness of chained guide trees. Bioinformatics 2016, 32, 3246–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Naser-Khdour, S.; Minh, B.Q.; Lanfear, R. Assessing confidence in root placement on phylogenies: An empirical study using non-reversible models for Mammals. Syst. Biol. 2021, syab067. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wooley, J.C.; Godzik, A. Probing metagenomics by rapid cluster analysis of very large datasets. PLoS ONE 2008, 3, e3375. [Google Scholar] [CrossRef] [Green Version]
- García, P.; Monjardín, C.; Martín, R.; Madera, C.; Soberón, N.; Garcia, E.; Meana, A.; Suárez, J.E. Isolation of new Stenotrophomonas bacteriophages and genomic characterization of temperate phage S1. Appl. Environ. Microbiol. 2008, 74, 7552–7560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adriaenssens, E.M.; Tolstoy, I.; Kropinski, A.M.; Ramsey, J. Create One New Genus (Menderavirus) including Three New Species (Caudovirales: Myoviridae). Code 2010.101B ICTV March 2021, MSL #36. 2020. Available online: https://ictv.global/proposal/MSL36/ (accessed on 13 December 2021).
- Sirotkin, K.; Cooley, W.; Runnels, J.; Snyder, L.R. A role in true-late gene expression for the T4 bacteriophage 5′ polynucleotide kinase 3′ phosphatase. J. Mol. Biol. 1978, 123, 221–233. [Google Scholar] [CrossRef]
- Casjens, S.R. The DNA-packaging nanomotor of tailed bacteriophages. Nat. Rev. Microbiol. 2011, 9, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Feiss, M. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 2008, 42, 647–681. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.; São-José, C.; Azeredo, J. Phage-derived peptidoglycan degrading enzymes: Challenges and future prospects for in vivo therapy. Viruses 2018, 10, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrov, V.M.; Ratnayaka, S.; Karam, J.D. Genetic insertions and diversification of the PolB-type DNA polymerase (gp43) of T4-related phages. J. Mol. Biol. 2010, 395, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.; Harman, A. Horizontal gene transfer of a Chlamydial tRNA-guanine transglycosylase gene to eukaryotic microbes. Mol. Phylogenet. Evol. 2016, 94, 392–396. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Mukherjee, P.; Roy, P. Genomic potential of Stenotrophomonas maltophilia in bioremediation with an assessment of its multifaceted role in our environment. Front. Microbiol. 2016, 7, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, J.; Mamat, U.; Abda, E.M.; Kirchhoff, L.; Streit, W.R.; Schaible, U.E.; Niemann, S.; Kohl, T.A. Analysis of phylogenetic variation of Stenotrophomonas maltophilia reveals human-specific branches. Front. Microbiol. 2018, 9, 806–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gröschel, M.I.; Meehan, C.J.; Barilar, I.; Diricks, M.; Gonzaga, A.; Steglich, M.; Conchillo-Solé, O.; Scherer, I.-C.; Mamat, U.; Luz, C.F.; et al. The phylogenetic landscape and nosocomial spread of the multidrug-resistant opportunist Stenotrophomonas maltophilia. Nat. Commun. 2020, 11, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Lira, F.; Berg, G.; Martínez, J.L. Double-face meets the bacterial world: The opportunistic pathogen Stenotrophomonas maltophilia. Front. Microbiol. 2017, 8, 2190–2204. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y.; Jiang, Y.; Wu, H.; Sun, W.; Huang, Y.P. Characterization and genomic analysis of ɸSHP3, a new transposable bacteriophage infecting Stenotrophomonas maltophilia. J. Virol. 2021, 95, e00019-21. [Google Scholar] [CrossRef] [PubMed]
- Adamek, M.; Overhage, J.; Bathe, S.; Winter, J.; Fischer, R.; Schwartz, T. Genotyping of environmental and clinical Stenotrophomonas maltophilia isolates and their pathogenic potential. PLoS ONE. 2011, 6, e27615. [Google Scholar] [CrossRef] [Green Version]
- Sybesma, W.; Zbinden, R.; Chanishvili, N.; Kutateladze, M.; Chkhotua, A.; Ujmajuridze, A.; Mehnert, U.; Kessler, T.M. Bacteriophages as potential treatment for urinary tract infections. Front. Microbiol. 2016, 7, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Gil-Gil, T.; Corona, F.; Martínez, J.L.; Bernardini, A. The Inactivation of enzymes belonging to the central carbon metabolism is a novel mechanism of developing antibiotic resistance. mSystems 2020, 5, e00282-20. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.B.; Hernandez, A.; Martinez, J.L. Stenotrophomonas maltophilia drug resistance. Future Microbiol. 2009, 4, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Dadonaite, B.; Vijayakrishnan, S.; Fodor, E.; Bhella, D.; Hutchinson, E.C. Filamentous influenza viruses. J. Gen. Virol. 2016, 97, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira-Garcia, D.; Dall’Agnol, M.; Rosales, M.; Azzuz, A.C.; Martinez, M.B.; Girón, J.A. Characterization of flagella produced by clinical strains of Stenotrophomonas maltophilia. Emerg. Infect. Dis. 2002, 8, 918–923. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Q.; Shen, P.; Huang, Y.P. Isolation and characterization of a novel filamentous phage from Stenotrophomonas maltophilia. Arch. Virol. 2012, 157, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Garza, K.D.; Newkirk, H.; Moreland, R.; Gonzalez, C.F.; Liu, M.; Ramsey, J.; Leavitt, J. Complete genome sequence of Stenotrophomonas phage Mendera. Microbiol. Resour. Announc. 2020, 9, e01411-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicary, A.; Newkirk, H.; Moreland, R.; Gonzalez, C.F.; Liu, M.; Ramsey, J.; Leavitt, J. Complete genome sequence of Stenotrophomonas maltophilia myophage Moby. Microbiol. Resour. Announc. 2020, 9, e01422-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshminarayan, R.; Phillips, B.P.; Binnian, I.L.; Gomez-Navarro, N.; Escudero-Urquijo, N.; Warren, A.J.; Miller, E.A. Pre-emptive quality control of a misfolded membrane protein by ribosome-driven effects. Curr. Biol. 2020, 30, 854–864. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Gao, M. Jumbo bacteriophages: An overview. Front. Microbiol. 2017, 8, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Li, Z.; Miller, E.S. Vibrio phage KVP40 encodes a functional NAD+ salvage pathway. J. Bacteriol. 2017, 199, e00855-16. [Google Scholar] [CrossRef] [Green Version]
- Decewicz, P.; Golec, P.; Szymczak, M.; Radlinska, M.; Dziewit, L. Identification and characterization of the first virulent phages, including a novel jumbo virus, infecting Ochrobactrum spp. Int. J. Mol. Sci. 2020, 21, 2096. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.M.; Arellano-Santoyo, H.; Temple, E.R.; Costliow, Z.A.; Pichaud, M.; Brantley Hall, A.; Liu, K.; Durney, M.A.; Gu, X.; Plichta, D.R.; et al. Gut microbiome ADP-ribosyltransferases are widespread phage-encoded fitness factors. Cell Host Microbe 2021, 29, 1351–1365. [Google Scholar] [CrossRef]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Summer, E.J.; Berry, J.; Tran, T.A.; Niu, L.; Struck, D.K.; Young, R. Rz/Rz1 lysis gene equivalents in phages of Gram-negative hosts. J. Mol. Biol. 2007, 373, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Albers, S.; Czech, A. Exploiting tRNAs to boost virulence. Life 2016, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgado, S.M.; Vicente, A.C.P. Mycobacterium genus and tRNA arrays. Mem. Inst. Oswaldo Cruz. 2019, 114, e180443. [Google Scholar] [CrossRef] [PubMed]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phage. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Yacoubi, B.; Bailly, M.; de Crécy-Lagard, V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012, 46, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Matuszek, Z.; Huang, Y.; Parisien, M.; Dai, Q.; Clark, W.; Schwartz, M.H.; Pan, T. Queuosine modification protects cognate tRNAs against ribonuclease cleavage. RNA 2018, 24, 1305–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinayak, M.; Pathak, C. Queuosine modification of tRNA: Its divergent role in cellular machinery. Biosci. Rep. 2009, 30, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Sabri, M.; Häuser, R.; Ouellette, M.; Liu, J.; Dehbi, M.; Moeck, G.; García, E.; Titz, B.; Uetz, P.; Moineau, S. Genome annotation and intraviral interactome for the Streptococcus pneumoniae virulent phage Dp-1. J. Bacteriol. 2011, 193, 551–562. [Google Scholar] [CrossRef] [Green Version]
- Holmfeldt, K.; Solonenko, N.; Shah, M.; Corrier, K.; Riemann, L.; Verberkmoes, N.C.; Sullivan, M.B. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc. Natl. Acad. Sci. USA 2013, 110, 12798–12803. [Google Scholar] [CrossRef] [Green Version]
- Sazinas, P.; Redgwell, T.; Rihtman, B.; Grigonyte, A.; Michniewski, S.; Scanlan, D.J.; Hobman, J.; Millard, A. Comparative genomics of bacteriophage of the genus Seuratvirus. Genome Biol. Evol. 2018, 10, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Hutinet, G.; Kot, W.; Cui, L.; Hillebrand, R.; Balamkundu, S.; Gnanakalai, S.; Neelakandan, R.; Carstens, A.B.; Lui, C.F.; Tremblay, D.; et al. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 2019, 10, 5442–5453. [Google Scholar] [CrossRef] [PubMed]
- Mackrill, J.J. Ryanodine receptor calcium release channels: An evolutionary perspective. Adv. Exp. Med. Biol. 2012, 740, 159–182. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Lewis, D.; des Georges, A.; Marks, A.R.; Frank, J. Ryanodine receptor structure and function in health and disease. Subcell. Biochem. 2018, 87, 329–352. [Google Scholar] [CrossRef] [Green Version]
- Ellenberger, T. Getting a grip on DNA recognition: Structures of the basic region leucine zipper, and the basic region helix-loop-helix DNA-binding domains. Curr. Opin. Struct. Biol. 1994, 4, 12–21. [Google Scholar] [CrossRef]
- Deppmann, C.D.; Alvania, R.S.; Taparowsky, E.J. Cross-species annotation of basic leucine zipper factor interactions: Insight into the evolution of closed interaction networks. Mol. Biol. Evol. 2006, 23, 1480–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clyde, K.; Glaunsinger, B.A. Chapter 1—Getting the message: Direct manipulation of host mRNA accumulation during Gammaherpesvirus lytic infection. In Advances in Virus Research; Maramorosch, K., Shatkin, A.J., Murphy, F.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; Volume 78, pp. 1–42. [Google Scholar] [CrossRef]
- Merrikh, C.N.; Merrikh, H. Gene inversion potentiates bacterial evolvability and virulence. Nat. Commun. 2018, 9, 4662–4671. [Google Scholar] [CrossRef] [PubMed]
- Rahimi-Midani, A.; Lee, Y.S.; Kang, S.W.; Kim, M.K.; Choi, T.J. First isolation and molecular characterization of bacteriophages infecting Acidovorax citrulli, the causal agent of bacterial fruit blotch. Plant Pathol. J. 2018, 34, 59–64. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Isolation Date | Isolation Source | Keratitis-Associated (Y/N) | Antibiotic Resistance * | MPDS Resistance # |
|---|---|---|---|---|---|
| Xmal2 | 4 February 1994 | Eye swab | N | azt (I), imi (R), tob (R), chl (I) | AQ, RH, MC |
| Xmal7 | 23 September 1994 | Eye swab | Y | azt (I), imi (R), chl (I) | AQ, RH, MC |
| Xmal10 | 15 December 1994 | Eye swab | Y | azt (I), imi (R), chl (I) | AQ, RH, EM, MC |
| Xmal12 | 12 October 1995 | Eye swab | Y | ND | ND |
| Xmal15 | 19 March 1998 | Contact lens | Y | azt (I), imi (R), chl (R) | AQ, RH, MC |
| Xmal21 | 2 April 2001 | Contact lens | Y | cip (R), ofl (I), imi (R), gen (R), tob (R), chl (I) | AQ, RH |
| Smal1 | 12 December 2005 | Cystic fibrosis swab | N | azt (I), imi (R), chl (I) | AQ, RH, MC |
| Smal2 | 25 May 1998 | Contact lens case | N | cip (I), imi (R), gen (I), chl (I) | AQ, RH, MC |
| Smal6 | 9 September 2010 | Contact lens case | Y | azt (R), cep (R), imi (R), chl (I) | AQ, RH, MC |
| Smal7 | 11 November 2010 | Contact lens case | N | azt (R), imi (R) | AQ, RH |
| Smal8 | 26 November 2010 | Contact lens case | N | imi (R), chl (I) | AQ, RH |
| Smal9 | 1 February 2011 | Contact lens case | N | imi (R) | AQ, RH, MC |
| Smal10 | 14 February 2011 | Contact lens case | N | imi (R) | AQ, RH, MC |
| Smal11 | 9 March 2011 | Contact lens case | N | azt (I), imi (R), pmb (R) | AQ, RH, EM, MC |
| Smal12 | 24 March 2011 | Contact lens case | Y | imi (R) | AQ, RH, MC |
| Smal13 | 1 April 2011 | Contact lens case | N | azt (R), cep (R), imi (R), | AQ, RH, MC |
| Smal14 | 5 April 2011 | Contact lens case | N | azt (I), imi (R), chl (R), | AQ, RH |
| Smal15 | 7 April 2011 | Contact lens case | N | azt (R), imi (R), chl (I) | AQ, RH, MC |
| Smal16 | 17 May 2011 | Contact lens case | N | azt (I), imi (R), | AQ, RH, MC |
| Smal17 | 6 June 2011 | Contact lens case | N | imi (R) | AQ, RH, MC |
| Smal18 | 1 July 2011 | Contact lens case | N | azt (R), cep (I), imi (R), | BT, AQ, RH, MC |
| Smal19 | 8 July 2011 | Contact lens case | N | azt (I), cep (R), imi (R) | BT, AQ, RH, MC |
| Smal20 | 5 July 2011 | Contact lens case | N | azt (R), cep (I), imi (R), tic (R), chl (I) | AQ, RH, MC |
| Smal21 | February 2012 | Contact lens case | N | ND | ND |
| Strain Name | Relative Efficiency of Plating | Single Plaque Description |
|---|---|---|
| Smal15 | 1.9 | clear |
| Smal16 | 1.8 | clear |
| Smal20 | 1.67 | clear |
| Xmal2 | 1.17 | clear |
| Smal2 | 1.08 | turbid |
| Xmal10 | 1 | clear |
| AP143S | 1 | clear |
| Smal12 | 0.91 | clear |
| Smal6 | 0.82 | clear |
| Smal11 | 0.8 | clear |
| Smal21 | 0.76 | clear |
| Xmal7 | 0.75 | turbid |
| Smal14 | 0.42 | turbid |
| Smal17 | 0.38 | clear |
| Smal8 | 0.28 | clear |
| Smal9 | 0.23 | clear |
| Smal10 | 0.11 | clear |
| Xmal15 | 0.06 | clear |
| Smal1 | 0.04 | clear |
| Smal7 * | 0.04 | turbid |
| Smal13 * | 0.03 | turbid |
| Smal18 | No single plaques (plaque spots at 105–108 PFU/mL) | - |
| Smal19 | No single plaques (plaque spots at 105–108 PFU/mL) | - |
| Xmal12 | - | - |
| Xmal21 | - | - |
| PAO1 | - | - |
| Phage Name | Size (bp) | GC% | Proteins a | Coding Density | tRNAs ab | Genome Similarity/Query Cover (%) c | Average Amino Acid Identity (%) d | Shared Proteins (%) |
|---|---|---|---|---|---|---|---|---|
| Ps15 | 161,350 | 54.2 | 275 | 93.86 | 24 | 100.0/100 | 100 | 276 (100) |
| BUCT608 | 160,122 | 54.2 | 270 (266) | 94.15 | 24 (20) | 98.62/92 | 93.90 | 221 (81.85) |
| IME-SM1 | 159,514 | 54.1 | 267 (202) | 92.88 | 24 (20) | 98.50/91 | 93.63 | 210 (78.65) |
| Marzo | 159,384 | 54.0 | 262 (268) | 93.65 | 24 (23) | 97.49/90.5 | 91.75 | 213 (81.30) |
| Mendera | 159,961 | 54.0 | 272 (286) | 94.05 | 24 (23) | 98.07/89.5 | 91.13 | 207 (76.10) |
| Moby | 159,365 | 54.1 | 268 (271) | 94.02 | 24 (23) | 94.73/91.5 | 91.98 | 215 (80.22) |
| YB07 | 159,862 | 54.1 | 269 (257) | 93.85 | 24 (0) | 98.45/90.5 | 93.47 | 220 (81.78) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damnjanović, D.; Vázquez-Campos, X.; Elliott, L.; Willcox, M.; Bridge, W.J. Characterisation of Bacteriophage vB_SmaM_Ps15 Infective to Stenotrophomonas maltophilia Clinical Ocular Isolates. Viruses 2022, 14, 709. https://doi.org/10.3390/v14040709
Damnjanović D, Vázquez-Campos X, Elliott L, Willcox M, Bridge WJ. Characterisation of Bacteriophage vB_SmaM_Ps15 Infective to Stenotrophomonas maltophilia Clinical Ocular Isolates. Viruses. 2022; 14(4):709. https://doi.org/10.3390/v14040709
Chicago/Turabian StyleDamnjanović, Dragica, Xabier Vázquez-Campos, Lisa Elliott, Mark Willcox, and Wallace J. Bridge. 2022. "Characterisation of Bacteriophage vB_SmaM_Ps15 Infective to Stenotrophomonas maltophilia Clinical Ocular Isolates" Viruses 14, no. 4: 709. https://doi.org/10.3390/v14040709
APA StyleDamnjanović, D., Vázquez-Campos, X., Elliott, L., Willcox, M., & Bridge, W. J. (2022). Characterisation of Bacteriophage vB_SmaM_Ps15 Infective to Stenotrophomonas maltophilia Clinical Ocular Isolates. Viruses, 14(4), 709. https://doi.org/10.3390/v14040709