
Distinctive High Expression of Antiretroviral APOBEC3 Protein in Mouse Germinal Center B Cells

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
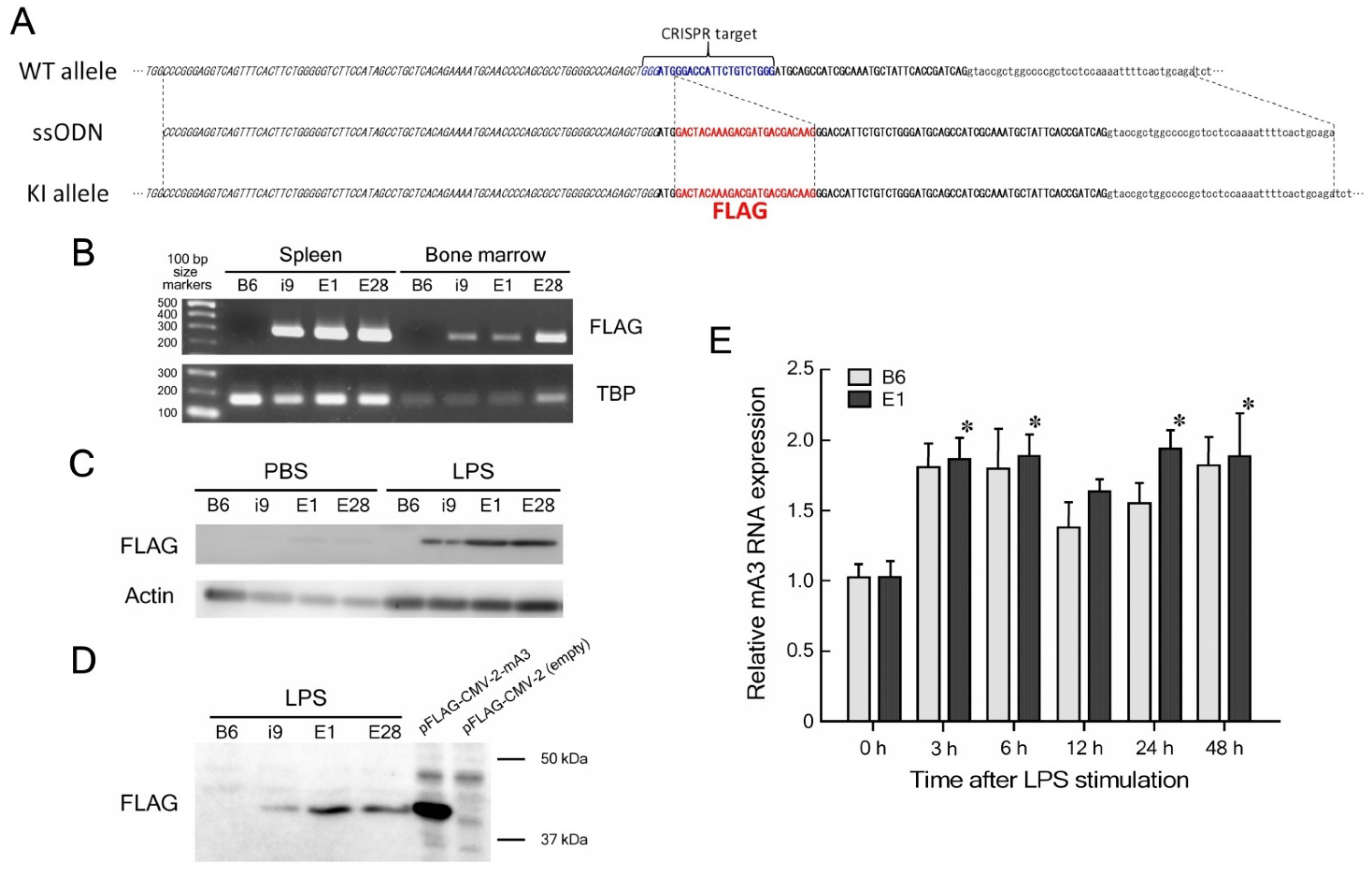
2.2. Generation of Apobec3-FLAG KI Mice
2.3. DNA Extraction and Genotyping
2.4. RNA Extraction, Reverse Transcription (RT)-PCR Assays and cDNA Sequencing
2.5. Quantitative Real-Time RT-PCR
2.6. Cell Culture
2.7. Western Blotting
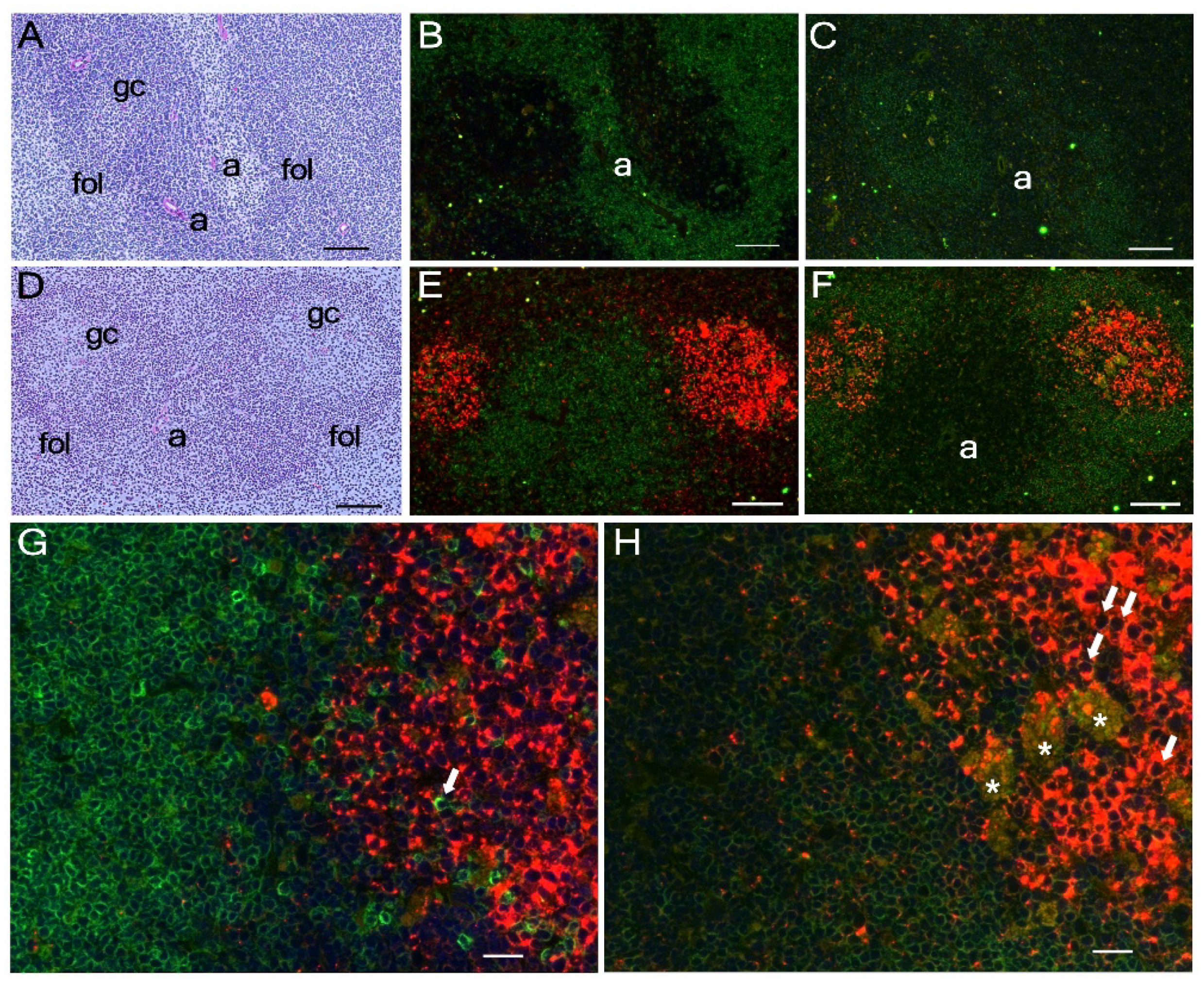
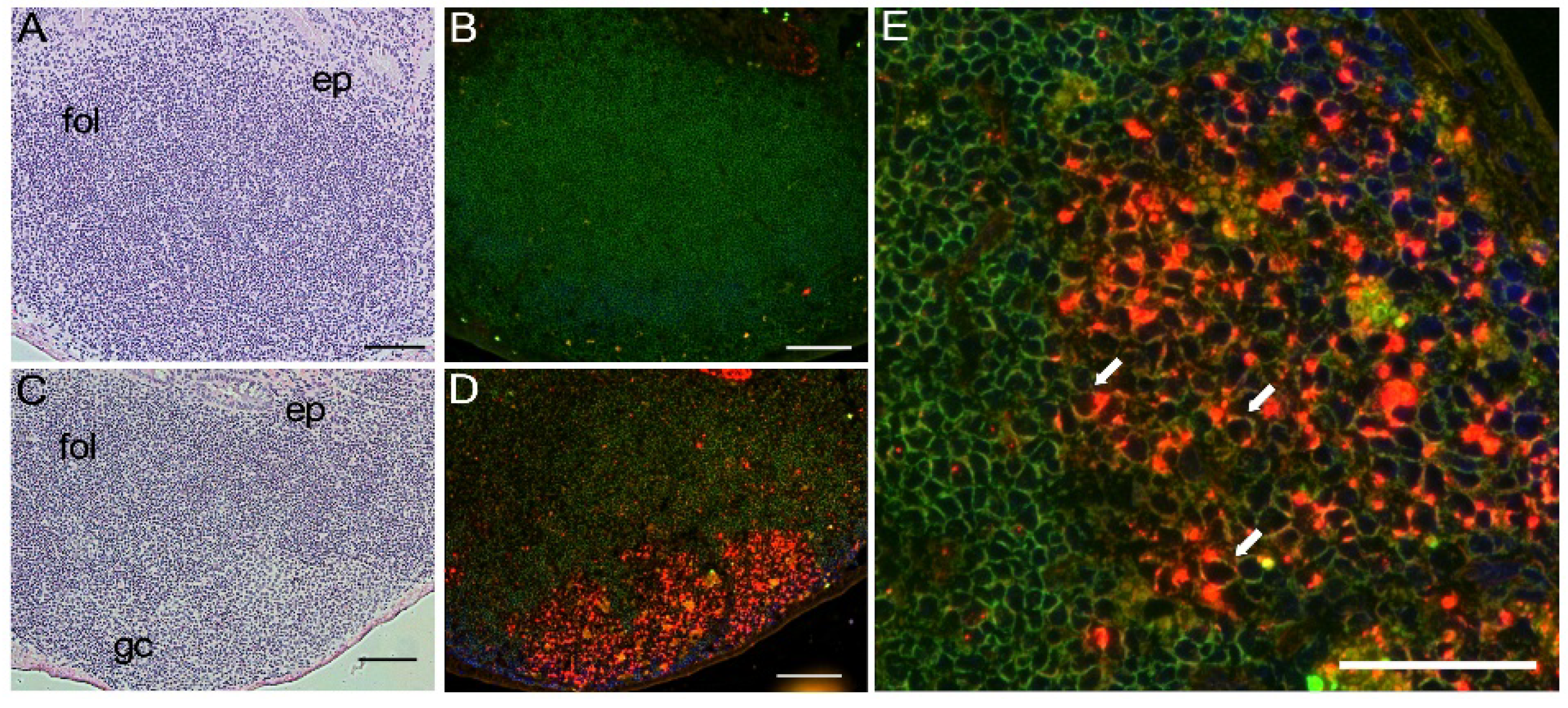
2.8. Immunohistochemistry
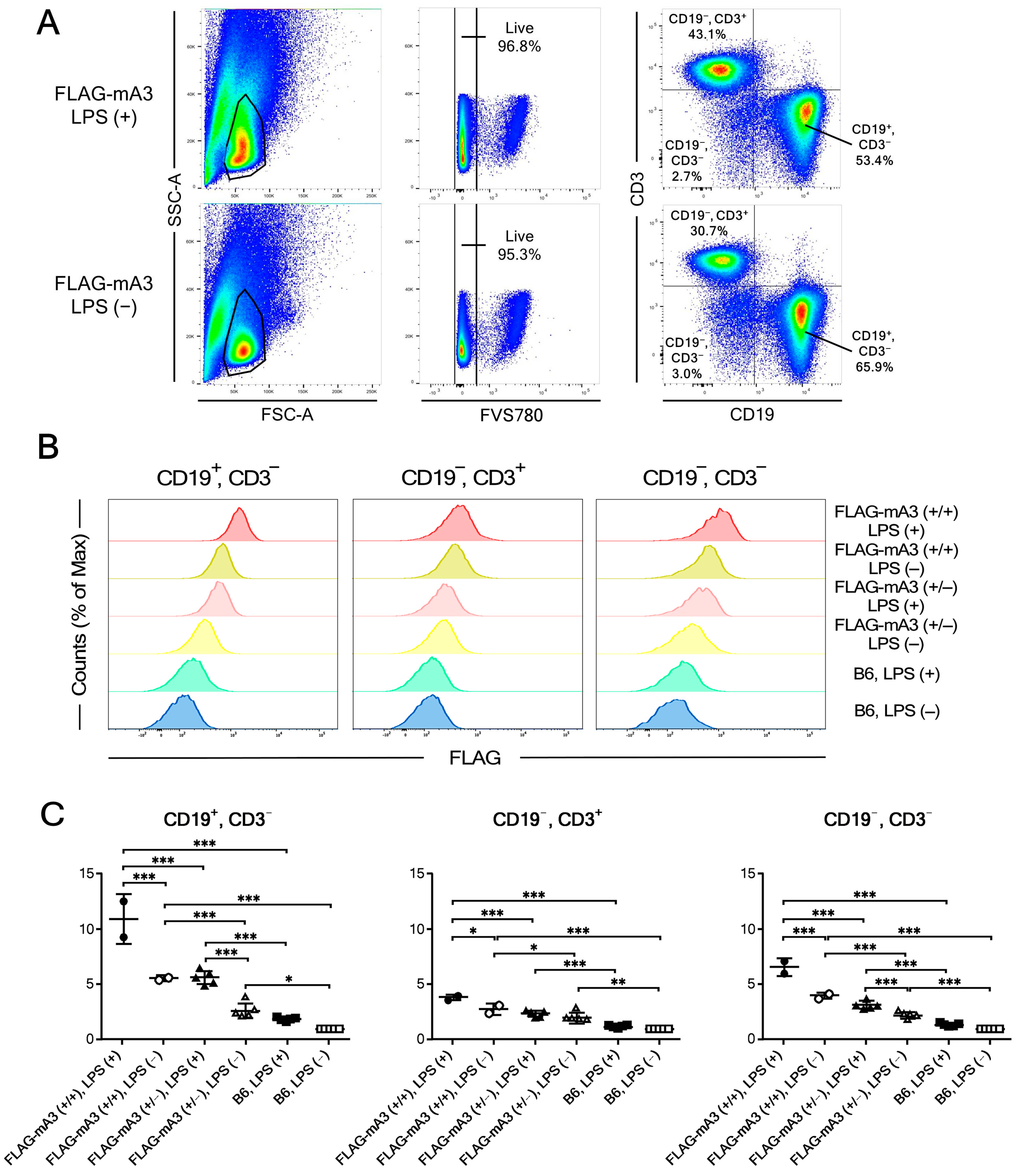
2.9. Flow Cytometry
2.10. Immunization with Sheep Red Blood Cells (SRBC)
2.11. Statistical Analyses
3. Results
3.1. Establishment of mA3 FLAG-KI Lines of Mice
3.2. Expression of mA3 Protein in LPS-Stimulated Spleen Cells
3.3. Tissue Distribution of mA3 Protein Expression In Vivo
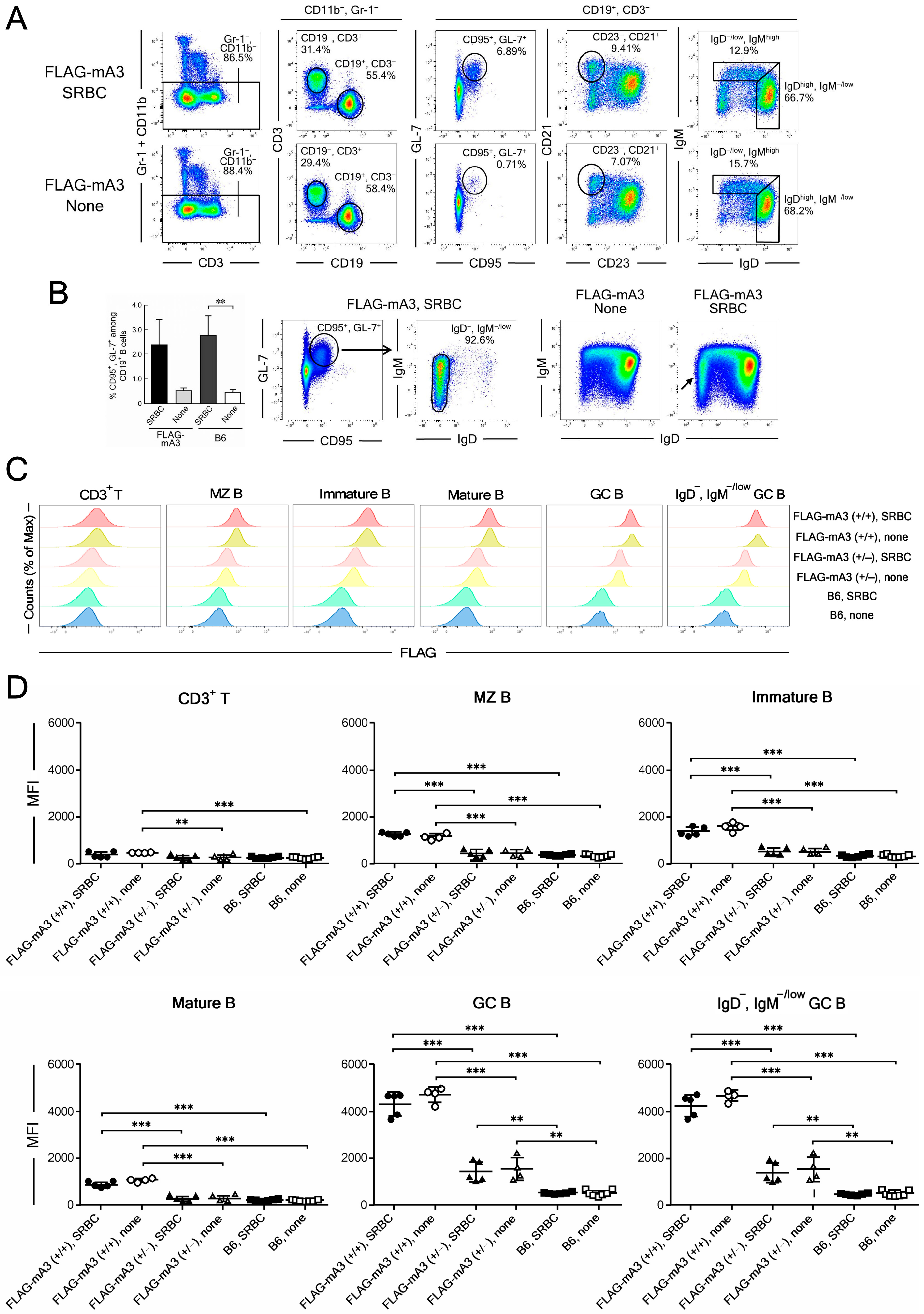
3.4. Flow Cytometric Analyses of B-Cell Subpopulations for Their mA3 Protein Expression before and after In Vivo Immunization with Sheep Red Blood Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Ito, J.; Gifford, R.J.; Sato, K. Retroviruses drive the rapid evolution of mammalian APOBEC3 genes. Proc. Nat. Acad. Sci. USA 2020, 117, 610–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uriu, K.; Kosugi, Y.; Ito, J.; Sato, K. The battle between retroviruses and APOBEC3 Genes: Its past and present. Viruses 2020, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Salas-Briceno, K.; Zhao, W.; Ross, S.R. Mouse APOBEC3 restriction of retroviruses. Viruses 2020, 12, 1217. [Google Scholar] [CrossRef] [PubMed]
- Hakata, Y.; Miyazawa, M. Deaminase-independent mode of antiretroviral action in human and mouse APOBEC3 proteins. Microorganisms 2020, 8, 1976. [Google Scholar] [CrossRef]
- Xu, W.K.; Byun, H.; Dudley, J.P. The role of APOBECs in viral replication. Microorganisms 2020, 8, 1899. [Google Scholar] [CrossRef]
- Sauter, D.; Kirchhoff, F. Key viral adaptations preceding the AIDS pandemic. Cell Host Microbe 2019, 25, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Okeoma, C.M.; Lovsin, N.; Peterlin, B.M.; Ross, S.R. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 2007, 445, 927–930. [Google Scholar] [CrossRef]
- Santiago, M.L.; Montano, M.; Benitez, R.; Messer, R.J.; Yonemoto, W.; Chesebro, B.; Hasenkrug, K.J.; Greene, W.C. Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science 2008, 321, 1343–1346. [Google Scholar] [CrossRef] [Green Version]
- Takeda, E.; Tsuji-Kawahara, S.; Sakamoto, M.; Langlois, M.-A.; Neuberger, M.S.; Rada, C.; Miyazawa, M. Mouse APOBEC3 restricts Friend leukemia virus infection and pathogenesis in vivo. J. Virol. 2008, 82, 10998–11008. [Google Scholar] [CrossRef] [Green Version]
- Langlois, M.-A.; Kemmerich, K.; Rada, C.; Neuberger, M.S. The AKV murine leukemia virus is restricted and hypermutated by mouse APOBEC3. J. Virol. 2009, 83, 11550–11559. [Google Scholar] [CrossRef] [Green Version]
- Okeoma, C.M.; Petersen, J.; Ross, S.R. Expression of murine APOBEC3 alleles in different mouse strains and their effect on mouse mammary tumor virus infection. J. Virol. 2009, 83, 3029–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanville, B.; Dolan, M.A.; Wollenberg, K.; Yan, Y.; Martin, C.; Yeung, M.L.; Strebel, K.; Buckler-White, A.; Kozak, C.A. Adaptive evolution of Mus Apobec3 includes retroviral insertion and positive selection at two clusters of residues flanking the substrate groove. PLoS Pathog. 2010, 6, e1000974. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hakata, Y.; Takeda, E.; Liu, Q.; Iwatani, Y.; Kozak, C.A.; Miyazawa, M. Two genetic determinants acquired late in Mus evolution regulate the inclusion of exon 5, which alters mouse APOBEC3 translation efficiency. PLoS Pathog. 2012, 8, e1002478. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Gracia, K.; Gramalla-Schmitz, A.; Weischedel, J.; Chahwan, R. APOBECs orchestrate genomic and epigenomic editing across health and disease. Trends Genet. 2021, 37, 1028–1043. [Google Scholar] [CrossRef]
- Henderson, S.; Fenton, T. APOBEC3 genes: Retroviral restriction factors to cancer drivers. Trends Mol. Med. 2015, 21, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, M.G.; Flath, B.; Chelico, L. The interesting relationship between APOBEC3 deoxycytidine deaminases and cancer: A long road ahead. Open Biol. 2020, 10, 200188. [Google Scholar] [CrossRef]
- Covino, D.A.; Gauzzi, M.C.; Fantuzzi, L. Understanding the regulation of APOBEC3 expression: Current evidence and much to learn. J. Leukoc. Biol. 2018, 103, 433–444. [Google Scholar] [CrossRef]
- OhAinle, M.; Kerns, J.A.; Li, M.M.H.; Malik, H.S.; Emerman, M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 2008, 4, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Ooms, M.; Brayton, B.; Letko, M.; Maio, S.M.; Pilcher, C.D.; Hecht, F.M.; Barbour, J.D.; Simon, V. HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe 2013, 14, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Seishima, N.; Kondo, S.; Wakae, K.; Wakisaka, N.; Kobayashi, E.; Kano, M.; Moriyama-Kita, M.; Nakanishi, Y.; Endo, K.; Imoto, T.; et al. Expression and subcellular localisation of AID and APOBEC3 in adenoid and palatine tonsils. Sci. Rep. 2018, 8, 918. [Google Scholar] [CrossRef]
- Koning, F.A.; Newman, E.N.C.; Kim, E.-Y.; Kunstman, K.J.; Wolinsky, S.M.; Malim, M.H. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009, 83, 9474–9485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, B.; Starrett, G.J.; Maurer, M.J.; Oberg, A.L.; Van Bockstal, M.; Van Dorpe, J.; De Wever, O.; Helleman, L.; Sieuwerts, A.M.; Berns, E.M.J.J.; et al. APOBEC3G expression correlates with T-cell infiltration and improved clinical outcomes in high-grade serous ovarian carcinoma. Clin. Cancer Res. 2016, 22, 4746–4755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.S.; Ruiz, A.; Gomez, L.M.; Miller, J.-M.; Berman, N.E.J.; Stephens, E.B. APOBEC3G expression is restricted to epithelial cells of the proximal convoluted tubules and is not expressed in the glomeruli of macaques. J. Histochem. Cytochem. 2007, 55, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okeoma, C.M.; Huegel, A.L.; Lingappa, J.; Feldman, M.D.; Ross, S.R. APOBEC3 proteins expressed in mammary epithelial cells are packaged into retroviruses and can restrict transmission of milk-borne virions. Cell Host Microbe 2010, 8, 534–543. [Google Scholar] [CrossRef] [Green Version]
- Dainichi, T.; Nakano, Y.; Wakae, K.; Otsuka, M.; Muramatsu, M.; Kabashima, K. APOBEC3 regulates keratinocyte differentiation and expression of Notch3. Exp. Dermatol. 2019, 28, 1341–1347. [Google Scholar] [CrossRef]
- Hakata, Y.; Li, J.; Fujino, T.; Tanaka, Y.; Shimizu, R.; Miyazawa, M. Mouse APOBEC3 interferes with autocatalytic cleavage of murine leukemia virus Pr180gag-pol precursor and inhibits Pr65gag processing. PLoS Pathog. 2019, 15, e1008173. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.L.; Law, E.K.; Argyris, P.P.; Carpenter, M.A.; Levin-Klein, R.; Ranum, A.N.; Molan, A.M.; Forster, C.L.; Anderson, B.D.; Lackey, L.; et al. A rabbit monoclonal antibody against the antiviral and cancer genomic DNA mutating enzyme APOBEC3B. Antibodies 2019, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Tsukaguchi, H.; Morita, H.; Higasa, K.; Tran, M.T.N.; Hamada, M.; Usui, T.; Morito, N.; Horita, S.; Hayashi, T.; et al. A mutation in transcription factor MAFB causes focal segmental glomerulosclerosis with Duane retraction syndrome. Kidney Int. 2018, 94, 396–407. [Google Scholar] [CrossRef]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- Mizuno, S.; Dinh, T.T.H.; Kato, K.; Mizuno-Iijima, S.; Tanimoto, Y.; Daitoku, Y.; Hoshino, Y.; Ikawa, M.; Takahashi, S.; Sugiyama, F.; et al. Simple generation of albino C57BL/6J mice with G291T mutation in the tyrosinase gene by the CRISPR/Cas9 system. Mamm. Genome 2014, 25, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Tsuji-Kawahara, S.; Chikaishi, T.; Takeda, E.; Kato, M.; Kinoshita, S.; Kajiwara, E.; Takamura, S.; Miyazawa, M. Persistence of viremia and production of neutralizing antibodies differentially regulated by polymorphic APOBEC3 and BAFF-R loci in Friend virus-infected mice. J. Virol. 2010, 84, 6082–6095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamura, S.; Tsuji-Kawahara, S.; Yagita, H.; Akiba, H.; Sakamoto, M.; Chikaishi, T.; Kato, M.; Miyazawa, M. Premature terminal exhaustion of Friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. J. Immunol. 2010, 184, 4696–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazawa, M.; Fujisawa, R.; Ishihara, C.; Takei, Y.A.; Shimizu, T.; Uenishi, H.; Yamagishi, H.; Kuribayashi, K. Immunization with a single T helper cell epitope abrogates Friend virus-induced early erythroid proliferation and prevents late leukemia development. J. Immunol. 1995, 155, 748–758. [Google Scholar] [PubMed]
- Takamura, S.; Kajiwara, E.; Tsuji-Kawahara, S.; Masumoto, T.; Fujisawa, M.; Kato, M.; Chikaishi, T.; Kawasaki, Y.; Kinoshita, S.; Itoi, M.; et al. Infection of adult thymus with murine retrovirus induces virus-specific central tolerance that prevents functional memory CD8+ T cell differentiation. PLoS Pathog. 2014, 10, e1003937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okeoma, C.M.; Low, A.; Bailis, W.; Fan, H.Y.; Peterlin, B.M.; Ross, S.R. Induction of APOBEC3 in vivo causes increased restriction of retrovirus infection. J. Virol. 2009, 83, 3486–3495. [Google Scholar] [CrossRef] [Green Version]
- Mehta, H.V.; Jones, P.H.; Weiss, J.P.; Okeoma, C.M. IFN-α and lipopolysaccharide upregulate APOBEC3 mRNA through different signaling pathways. J. Immunol. 2012, 189, 4088–4103. [Google Scholar] [CrossRef] [Green Version]
- McAllister, E.J.; Apgar, J.R.; Leung, C.R.; Rickert, R.C.; Jellusova, J. New methods to analyze B cell immune responses to thymus-dependent antigen sheep red blood cells. J. Immunol. 2017, 199, 2998–3003. [Google Scholar] [CrossRef] [Green Version]
- Shinall, S.M.; Gonzalez-Fernandez, M.; Noelle, R.J.; Waldschmidt, T.J. Identification of murine germinal center B cell subsets defined by the expression of surface isotypes and differentiation antigens. J. Immunol. 2000, 164, 5729–5738. [Google Scholar] [CrossRef] [Green Version]
- Roco, J.A.; Mesin, L.; Binder, S.C.; Nefzger, C.; Gonzalez-Figueroa, P.; Canete, P.F.; Ellyard, J.; Shen, Q.; Robert, P.A.; Cappello, J.; et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 2019, 51, 337–350. [Google Scholar] [CrossRef]
- Sharma, S.; Wang, J.; Alqassim, E.; Portwood, S.; Gomez, E.C.; Maguire, O.; Basse, P.H.; Wang, E.S.; Segal, B.H.; Baysal, B.E. Mitochondrial hypoxic stress induces widespread RNA editing by APOBEC3G in natural killer cells. Genome Biol. 2019, 20, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanevskiy, L.M.; Telford, W.G.; Sapozhnikov, A.M.; Kovalenko, E.I. Lipopolysaccharide induces IFN-γ production in human NK cells. Front. Immunol. 2013, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasenkrug, K.J.; Brooks, D.M.; Dittmer, U. Critical role for CD4+ T cells in controlling retrovirus replication and spread in persistently infected mice. J. Virol. 1998, 72, 6559–6564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windmann, S.; Otto, L.; Hrycak, C.P.; Malyshkina, A.; Bongard, N.; David, P.; Gunzer, M.; Dittmer, U.; Bayer, W. Infection of B cell follicle-resident cells by Friend retrovirus occurs during acute infection and is maintained during viral persistence. mBio 2019, 10, e00004-19. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Tsuji-Kawahara, S.; Kawasaki, Y.; Kinoshita, S.; Chikaishi, T.; Takamura, S.; Fujisawa, M.; Kawada, A.; Miyazawa, M. Class switch recombination and somatic hypermutation of virus-neutralizing antibodies are not essential for the control of Friend retrovirus infection. J. Virol. 2015, 89, 1468–1473. [Google Scholar] [CrossRef] [Green Version]
- Halemano, K.; Guo, K.; Heilman, K.J.; Barrett, B.S.; Smith, D.S.; Hasenkrug, K.J.; Santiago, M.L. Immunoglobulin somatic hypermutation by APOBEC3/Rfv3 during retroviral infection. Proc. Natl. Acad. Sci. USA 2014, 111, 7759–7764. [Google Scholar] [CrossRef] [Green Version]
- Lackey, L.; Law, E.K.; Brown, W.L.; Harris, R.S. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle 2013, 12, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Lackey, L.; Demorest, Z.L.; Land, A.M.; Hultquist, J.F.; Brown, W.L.; Harris, R.S. APOBEC3B and AID have similar nuclear import mechanisms. J. Mol. Biol. 2012, 419, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Willenbrock, K.; Jungnickel, B.; Hansmann, M.-L.; Küppers, R. Human splenic marginal zone B cells lack expression of activation-induced cytidine deaminase. Eur. J. Immunol. 2005, 35, 3002–3007. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsukimoto, S.; Hakata, Y.; Tsuji-Kawahara, S.; Enya, T.; Tsukamoto, T.; Mizuno, S.; Takahashi, S.; Nakao, S.; Miyazawa, M. Distinctive High Expression of Antiretroviral APOBEC3 Protein in Mouse Germinal Center B Cells. Viruses 2022, 14, 832. https://doi.org/10.3390/v14040832
Tsukimoto S, Hakata Y, Tsuji-Kawahara S, Enya T, Tsukamoto T, Mizuno S, Takahashi S, Nakao S, Miyazawa M. Distinctive High Expression of Antiretroviral APOBEC3 Protein in Mouse Germinal Center B Cells. Viruses. 2022; 14(4):832. https://doi.org/10.3390/v14040832
Chicago/Turabian StyleTsukimoto, Shota, Yoshiyuki Hakata, Sachiyo Tsuji-Kawahara, Takuji Enya, Tetsuo Tsukamoto, Seiya Mizuno, Satoru Takahashi, Shinichi Nakao, and Masaaki Miyazawa. 2022. "Distinctive High Expression of Antiretroviral APOBEC3 Protein in Mouse Germinal Center B Cells" Viruses 14, no. 4: 832. https://doi.org/10.3390/v14040832
APA StyleTsukimoto, S., Hakata, Y., Tsuji-Kawahara, S., Enya, T., Tsukamoto, T., Mizuno, S., Takahashi, S., Nakao, S., & Miyazawa, M. (2022). Distinctive High Expression of Antiretroviral APOBEC3 Protein in Mouse Germinal Center B Cells. Viruses, 14(4), 832. https://doi.org/10.3390/v14040832