
Foot-and-Mouth Disease Virus 3A Hijacks Sar1 and Sec12 for ER Remodeling in a COPII-Independent Manner


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Plasmids
2.2.1. 3A-Associated Constructs
2.2.2. Host-Factor Plasmids
2.2.3. Others
2.3. Monoclonal Antibody Preparation
2.4. Western Blotting
2.5. Immunofluorescence Assay and Confocal Live-Cell Imaging
2.6. Autophagy Induction
2.7. Transmission Electron Microscopy and Electron Tomography
2.8. Recombinant Vaccinia Expression System
2.9. Immunoprecipitation Assay
2.10. Knockdown Assay
3. Results
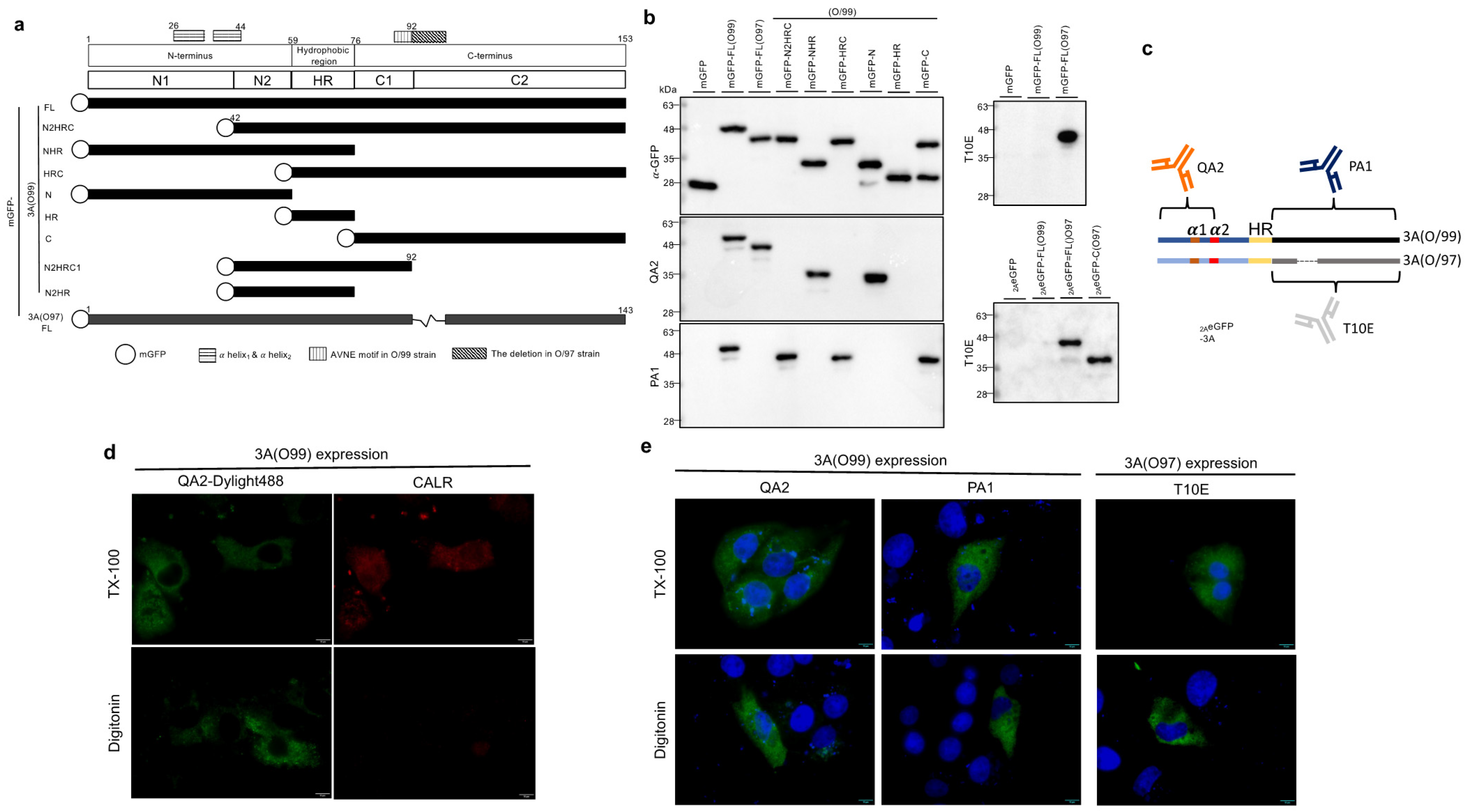
3.1. Membrane Topology of Peripheral FMDV 3A Protein
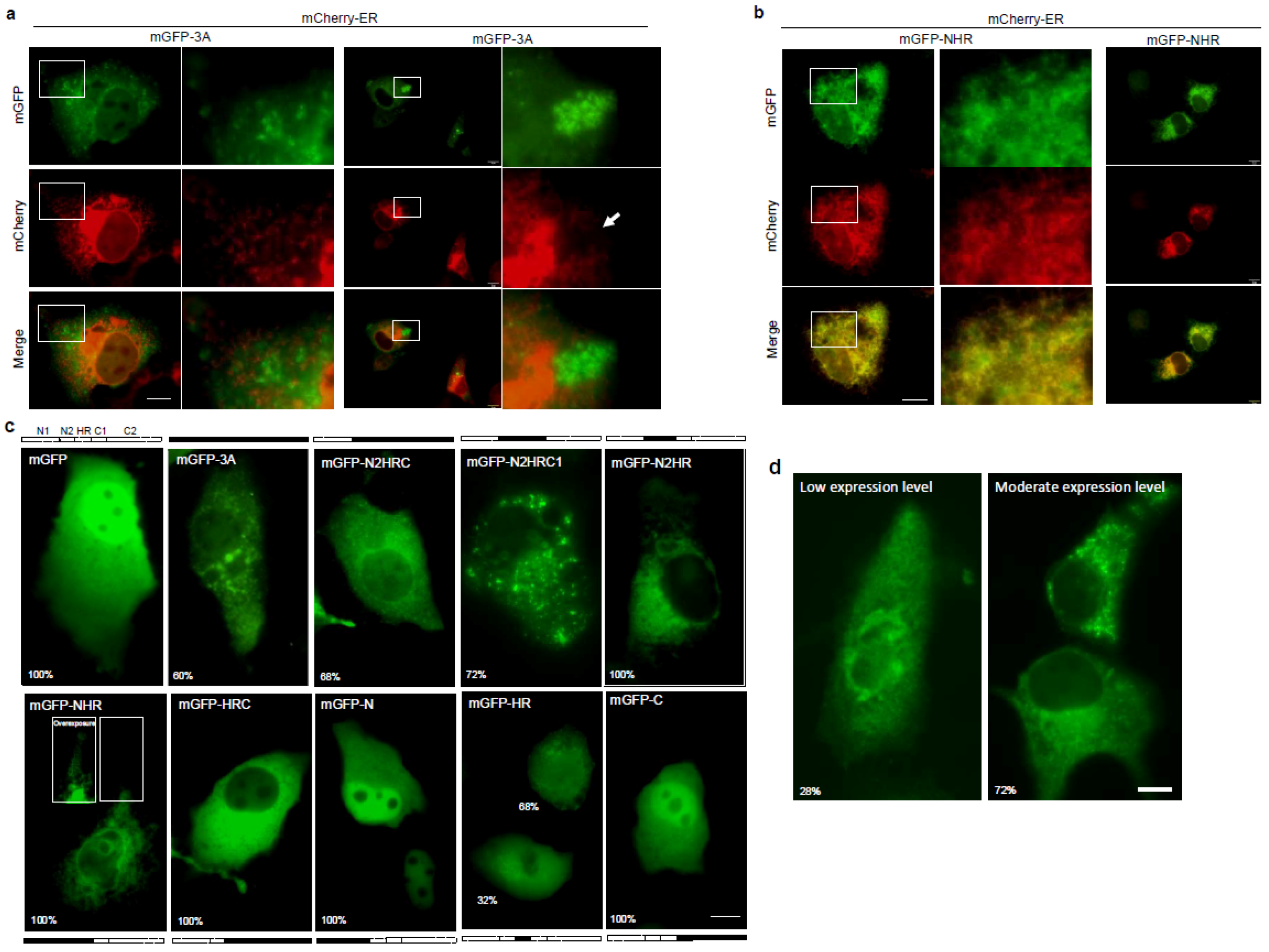
3.2. FMDV 3A Protein Modified ER into Punctae
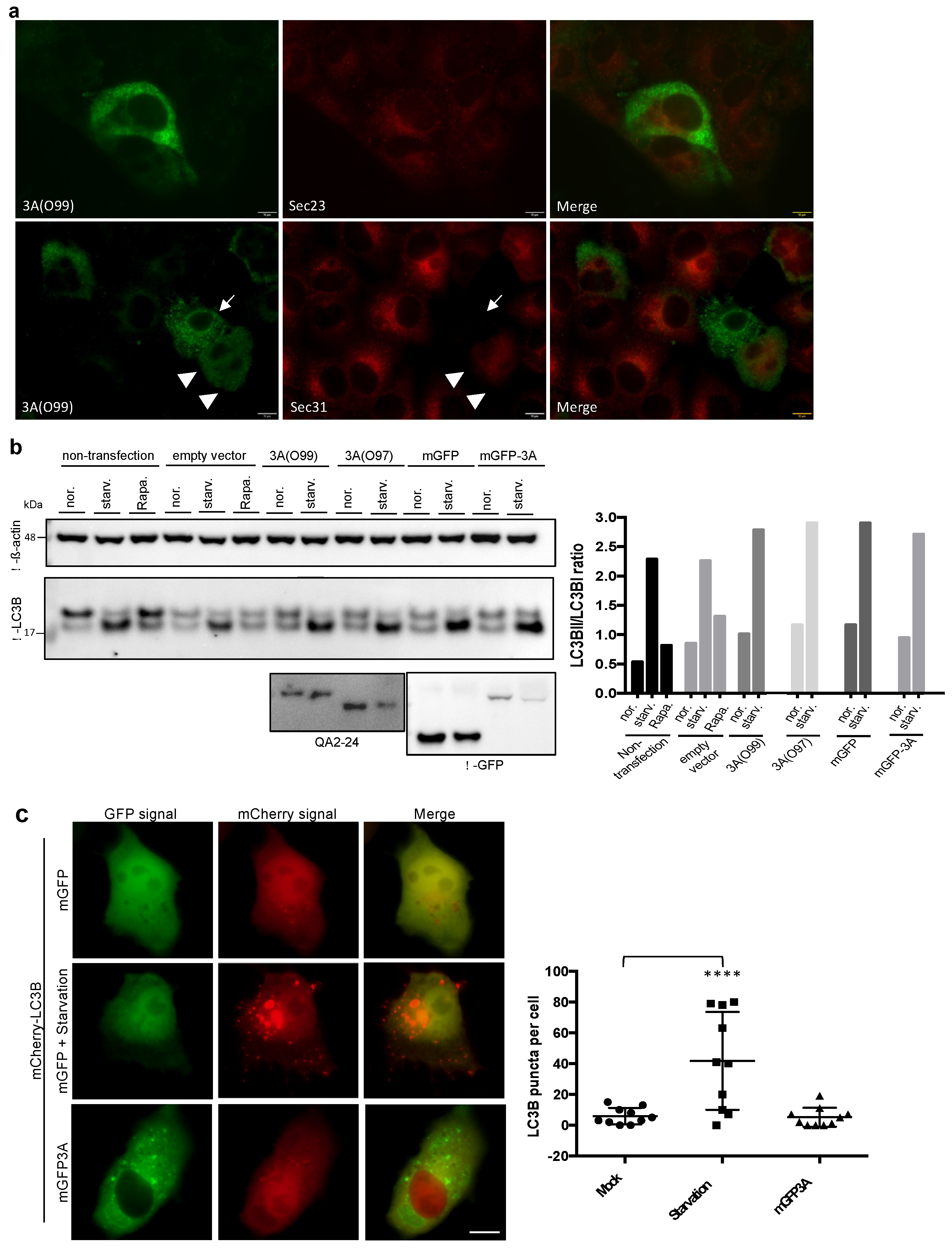
3.3. The 3A Punctae Were Distinct from Traditional COPII Vesicles or Autophagosomes
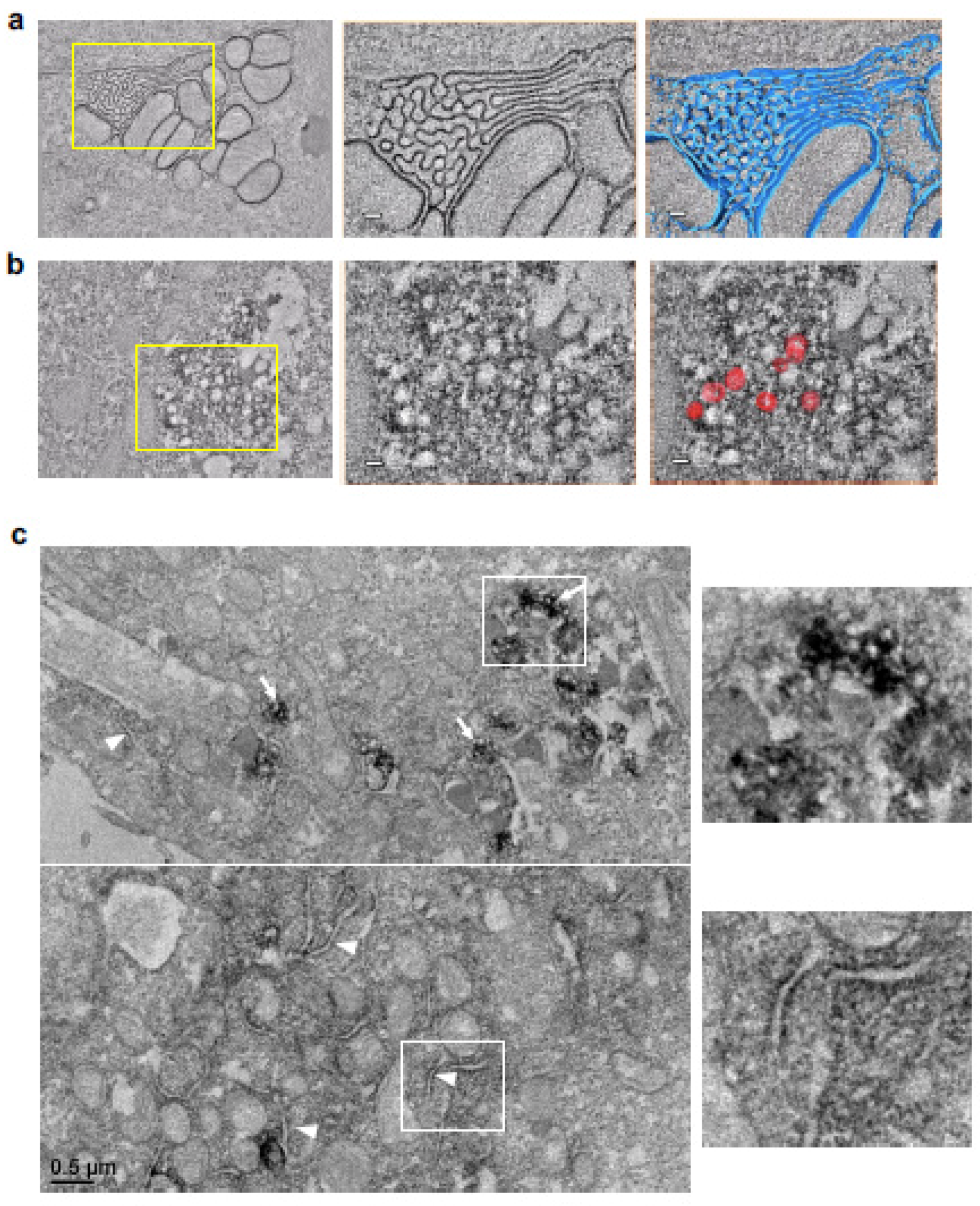
3.4. The Ultrastructure of the 3A Vesicle-like Structure as Observed Using Transmission Electron Microscopy
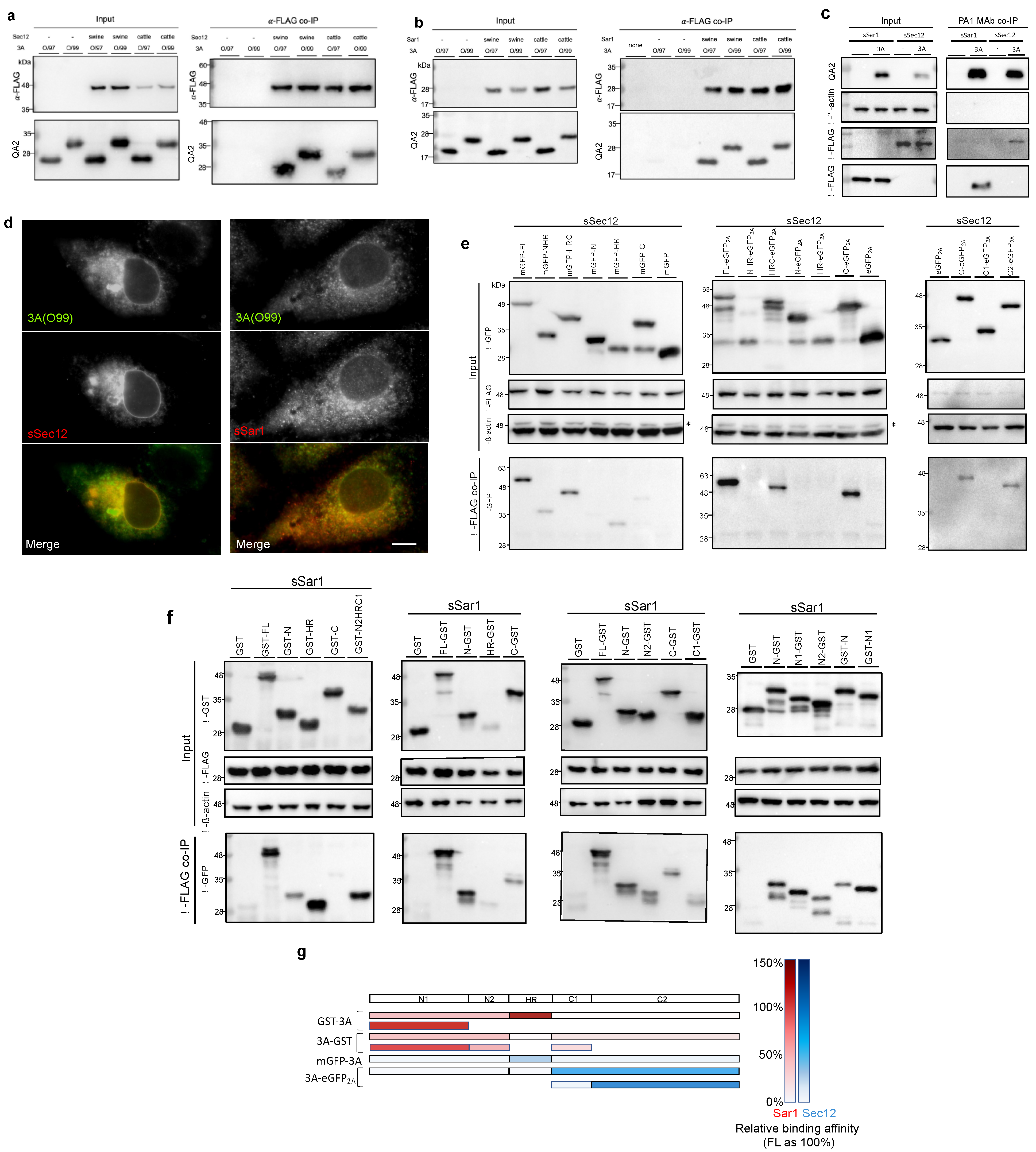
3.5. COPII Factors, Sar1 and Sec12, Were Found to Be Novel 3A Interaction Partners
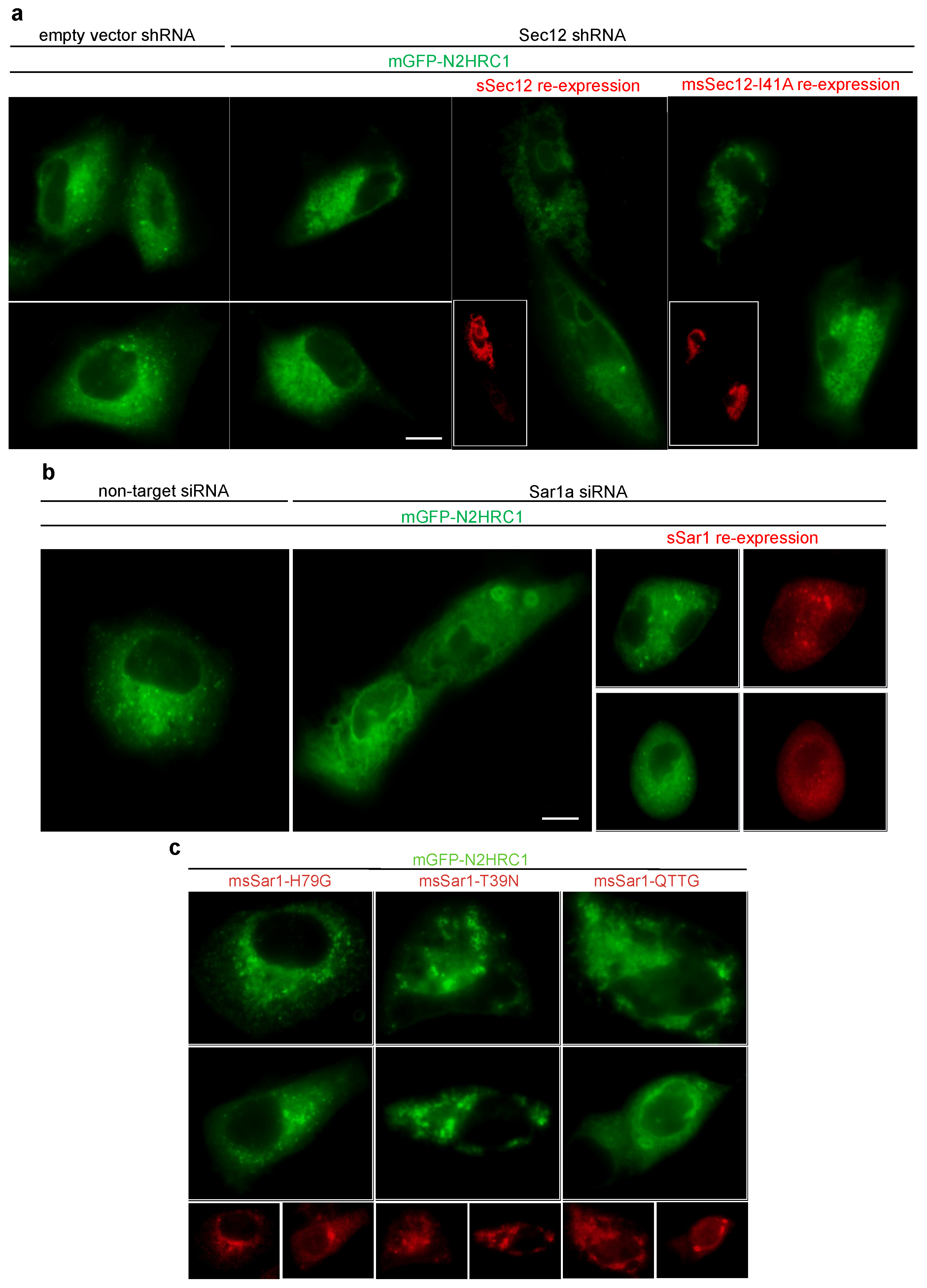
3.6. Knockdown of Sar1 or Sec12 Inhibited Formation of 3A Punctae
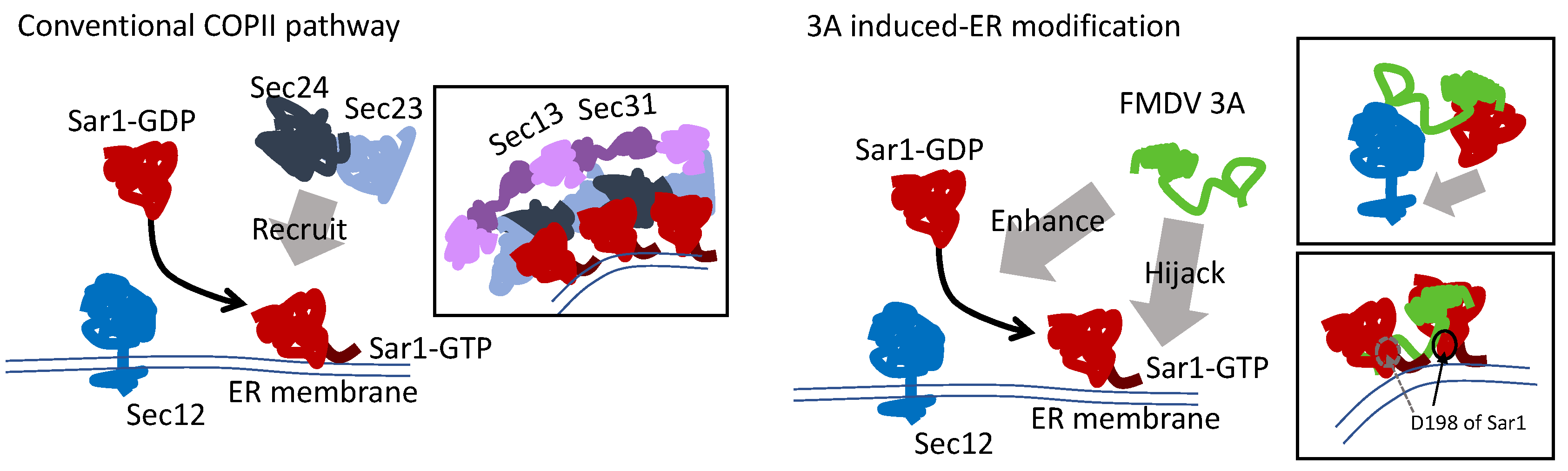
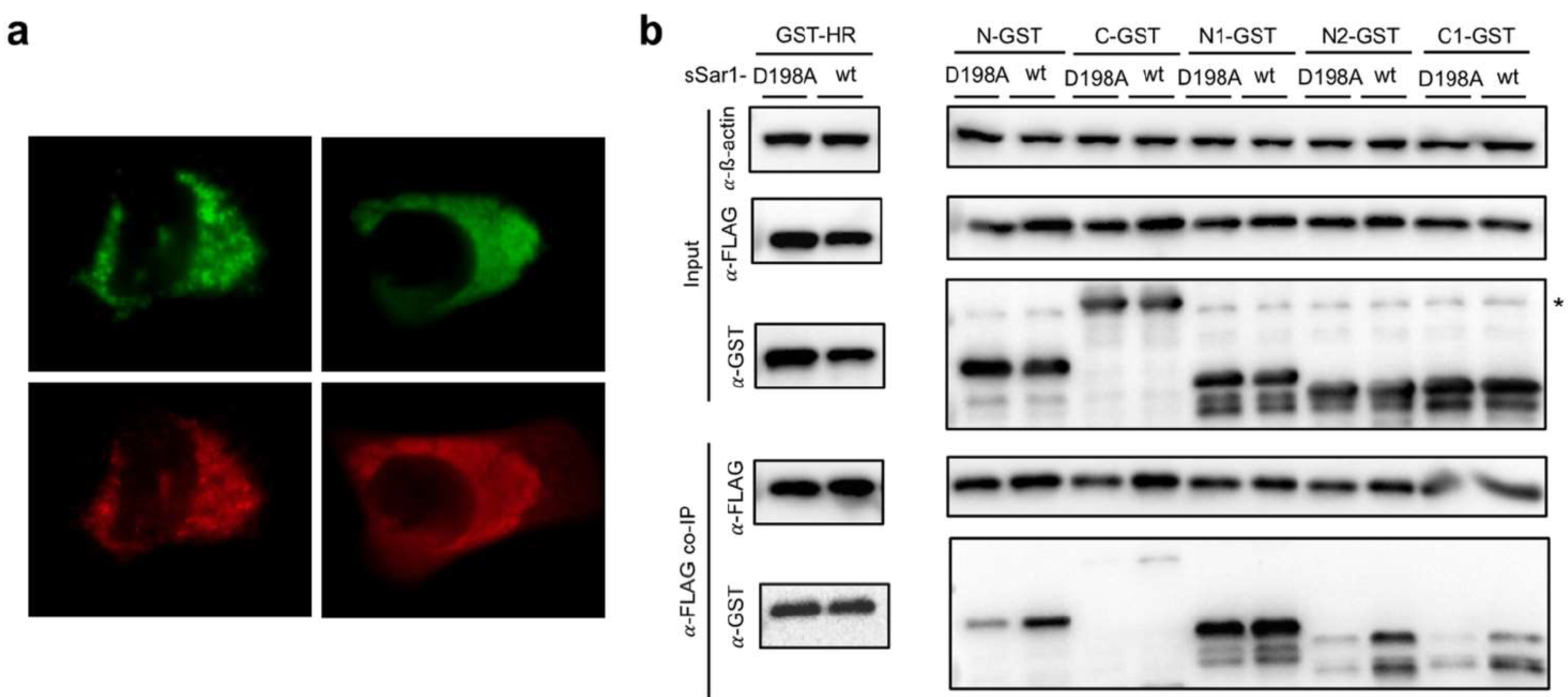
3.7. The Model of Active Sar1–3A Intercalating Complex for ER Remodeling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Belov, G.A.; van Kuppeveld, F.J. (+)RNA viruses rewire cellular pathways to build replication organelles. Curr. Opin. Virol. 2012, 2, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, P.; Cook, H.; Jackson, T.; Ryan, M.; Wileman, T. The ultrastructure of the developing replication site in foot-and-mouth disease virus-infected BHK-38 cells. J. Gen. Virol. 2004, 85 Pt 4, 933–946. [Google Scholar] [CrossRef] [PubMed]
- van der Schaar, H.M.; Dorobantu, C.M.; Albulescu, L.; Strating, J.; van Kuppeveld, F.J.M. Fat(al) attraction: Picornaviruses Usurp Lipid Transfer at Membrane Contact Sites to Create Replication Organelles. Trends Microbiol. 2016, 24, 535–546. [Google Scholar] [CrossRef]
- Greninger, A.L. Picornavirus-Host Interactions to Construct Viral Secretory Membranes. Mol. Basis Viral Infect. 2015, 129, 189–212. [Google Scholar]
- Egger, D.; Pasamontes, L.; Bolten, R.; Boyko, V.; Bienz, K. Reversible dissociation of the poliovirus replication complex: Functions and interactions of its components in viral RNA synthesis. J. Virol. 1996, 70, 8675–8683. [Google Scholar] [CrossRef]
- Wessels, E.; Duijsings, D.; Niu, T.K.; Neumann, S.; Oorschot, V.M.; de Lange, F.; Lanke, K.H.; Klumperman, J.; Henke, A.; Jackson, C.L.; et al. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev. Cell 2006, 11, 191–201. [Google Scholar] [CrossRef]
- Wessels, E.; Duijsings, D.; Notebaart, R.A.; Melchers, W.J.; van Kuppeveld, F.J. A proline-rich region in the coxsackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum-to-golgi transport. J. Virol. 2005, 79, 5163–5173. [Google Scholar] [CrossRef]
- Moghimi, S.; Viktorova, E.; Zimina, A.; Szul, T.; Sztul, E.; Belov, G.A. Enterovirus Infection Induces Massive Recruitment of All Isoforms of Small Cellular Arf GTPases to the Replication Organelles. J. Virol. 2020, 95, e01629-20. [Google Scholar] [CrossRef]
- Viktorova, E.G.; Gabaglio, S.; Meissner, J.M.; Lee, E.; Moghimi, S.; Sztul, E.; Belov, G.A. A Redundant Mechanism of Recruitment Underlies the Remarkable Plasticity of the Requirement of Poliovirus Replication for the Cellular ArfGEF GBF1. J. Virol. 2019, 93, e00856-19. [Google Scholar] [CrossRef]
- Greninger, A.L.; Knudsen, G.M.; Betegon, M.; Burlingame, A.L.; Derisi, J.L. The 3A protein from multiple picornaviruses utilizes the golgi adaptor protein ACBD3 to recruit PI4KIIIbeta. J. Virol. 2012, 86, 3605–3616. [Google Scholar] [CrossRef]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Shengjuler, D.; Chan, Y.M.; Sun, S.; Moustafa, I.M.; Li, Z.L.; Gohara, D.W.; Buck, M.; Cremer, P.S.; Boehr, D.D.; Cameron, C.E. The RNA-Binding Site of Poliovirus 3C Protein Doubles as a Phosphoinositide-Binding Domain. Structure 2017, 25, 1875–1886.e7. [Google Scholar] [CrossRef] [PubMed]
- Ilnytska, O.; Santiana, M.; Hsu, N.Y.; Du, W.L.; Chen, Y.H.; Viktorova, E.G.; Belov, G.; Brinker, A.; Storch, J.; Moore, C.; et al. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 2013, 14, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef]
- O’Donnell, V.K.; Pacheco, J.M.; Henry, T.M.; Mason, P.W. Subcellular distribution of the foot-and-mouth disease virus 3A protein in cells infected with viruses encoding wild-type and bovine-attenuated forms of 3A. Virology 2001, 287, 151–162. [Google Scholar] [CrossRef]
- Midgley, R.; Moffat, K.; Berryman, S.; Hawes, P.; Simpson, J.; Fullen, D.; Stephens, D.J.; Burman, A.; Jackson, T. A role for endoplasmic reticulum exit sites in foot-and-mouth disease virus infection. J. Gen. Virol. 2013, 94 Pt 12, 2636–2646. [Google Scholar] [CrossRef]
- Moffat, K.; Howell, G.; Knox, C.; Belsham, G.J.; Monaghan, P.; Ryan, M.D.; Wileman, T. Effects of foot-and-mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum-to-Golgi transport. J. Virol. 2005, 79, 4382–4395. [Google Scholar] [CrossRef]
- Moffat, K.; Knox, C.; Howell, G.; Clark, S.J.; Yang, H.; Belsham, G.J.; Ryan, M.; Wileman, T. Inhibition of the secretory pathway by foot-and-mouth disease virus 2BC protein is reproduced by coexpression of 2B with 2C, and the site of inhibition is determined by the subcellular location of 2C. J. Virol. 2007, 81, 1129–1139. [Google Scholar] [CrossRef]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef]
- Berryman, S.; Moffat, K.; Harak, C.; Lohmann, V.; Jackson, T. Foot-and-mouth disease virus replicates independently of phosphatidylinositol 4-phosphate and type III phosphatidylinositol 4-kinases. J. Gen. Virol. 2016, 97, 1841–1852. [Google Scholar] [CrossRef]
- Loundras, E.A.; Herod, M.R.; Harris, M.; Stonehouse, N.J. Foot-and-mouth disease virus genome replication is unaffected by inhibition of type III phosphatidylinositol-4-kinases. J. Gen. Virol. 2016, 97, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Seki, F.; Ono, N.; Yamaguchi, R.; Yanagi, Y. Efficient isolation of wild strains of canine distemper virus in Vero cells expressing canine SLAM (CD150) and their adaptability to marmoset B95a cells. J. Virol. 2003, 77, 9943–9950. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Magaldi, M.; Martin-Acebes, M.A.; Kremer, L.; Sobrino, F. Membrane topology and cellular dynamics of foot-and-mouth disease virus 3A protein. PLoS ONE 2014, 9, e106685. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Magaldi, M.; Postigo, R.; de la Torre, B.G.; Vieira, Y.A.; Rodriguez-Pulido, M.; Lopez-Vinas, E.; Gomez-Puertas, P.; Andreu, D.; Kremer, L.; Rosas, M.F.; et al. Mutations that hamper dimerization of foot-and-mouth disease virus 3A protein are detrimental for infectivity. J. Virol. 2012, 86, 11013–11023. [Google Scholar] [CrossRef] [PubMed]
- Beard, C.W.; Mason, P.W. Genetic determinants of altered virulence of Taiwanese foot-and-mouth disease virus. J. Virol. 2000, 74, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.M.; Gladue, D.P.; Holinka, L.G.; Arzt, J.; Bishop, E.; Smoliga, G.; Pauszek, S.J.; Bracht, A.J.; O’Donnell, V.; Fernandez-Sainz, I.; et al. A partial deletion in non-structural protein 3A can attenuate foot-and-mouth disease virus in cattle. Virology 2013, 446, 260–267. [Google Scholar] [CrossRef]
- Yang, W.; Li, D.; Ru, Y.; Bai, J.; Ren, J.; Zhang, J.; Li, L.; Liu, X.; Zheng, H. Foot-and-Mouth Disease Virus 3A Protein Causes Upregulation of Autophagy-Related Protein LRRC25 To Inhibit the G3BP1-Mediated RIG-Like Helicase-Signaling Pathway. J. Virol. 2020, 94, e02086-19. [Google Scholar] [CrossRef]
- Fu, S.Z.; Yang, W.P.; Ru, Y.; Zhang, K.S.; Wang, Y.; Liu, X.T.; Li, D.; Zheng, H.X. DDX56 cooperates with FMDV 3A to enhance FMDV replication by inhibiting the phosphorylation of IRF3. Cell Signal. 2019, 64, 109393. [Google Scholar] [CrossRef]
- Lawrence, P.; Rieder, E. Identification of RNA helicase A as a new host factor in the replication cycle of foot-and-mouth disease virus. J. Virol. 2009, 83, 11356–11366. [Google Scholar] [CrossRef]
- Ma, X.; Ling, Y.; Li, P.; Sun, P.; Cao, Y.; Bai, X.; Li, K.; Fu, Y.; Zhang, J.; Li, D.; et al. Cellular Vimentin Interacts with Foot-and-Mouth Disease Virus Nonstructural Protein 3A and Negatively Modulates Viral Replication. J. Virol. 2020, 94, e00273-20. [Google Scholar] [CrossRef]
- Gladue, D.P.; O’Donnell, V.; Baker-Bransetter, R.; Pacheco, J.M.; Holinka, L.G.; Arzt, J.; Pauszek, S.; Fernandez-Sainz, I.; Fletcher, P.; Brocchi, E.; et al. Interaction of foot-and-mouth disease virus nonstructural protein 3A with host protein DCTN3 is important for viral virulence in cattle. J. Virol. 2014, 88, 2737–2747. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, G.; Pahuja, K.B.; Studer, S.; Shim, S.; Schekman, R. COPII and the regulation of protein sorting in mammals. Nat. Cell Biol. 2011, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Gurkan, C.; Stagg, S.M.; Lapointe, P.; Balch, W.E. The COPII cage: Unifying principles of vesicle coat assembly. Nat. Rev. Mol. Cell Biol. 2006, 7, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef]
- Lee, H.W.; Deng, M.C.; Pan, C.H.; Chang, H.W.; Cheng, I.C. Neutralizing monoclonal antibodies against porcinophilic foot-and-mouth disease virus mapped to antigenic site 2 by utilizing novel mutagenic virus-like particles to detect the antigenic change. Vet. Microbiol. 2018, 222, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.F.; Lin, H.L.; Fu, C.Y. 3D Mitochondrial Ultrastructure of Drosophila Indirect Flight Muscle Revealed by Serial-section Electron Tomography. J. Vis. Exp. 2017, 130, 56567. [Google Scholar] [CrossRef]
- Fuerst, T.R.; Niles, E.G.; Studier, F.W.; Moss, B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 1986, 83, 8122–8126. [Google Scholar] [CrossRef]
- Choe, S.S.; Kirkegaard, K. Intracellular topology and epitope shielding of poliovirus 3A protein. J. Virol. 2004, 78, 5973–5982. [Google Scholar] [CrossRef]
- Roderick, H.L.; Campbell, A.K.; Llewellyn, D.H. Nuclear localisation of calreticulin in vivo is enhanced by its interaction with glucocorticoid receptors. FEBS Lett. 1997, 405, 181–185. [Google Scholar] [CrossRef]
- Tenorio, R.; Fernandez de Castro, I.; Knowlton, J.J.; Zamora, P.F.; Lee, C.H.; Mainou, B.A.; Dermody, T.S.; Risco, C. Reovirus sigmaNS and muNS Proteins Remodel the Endoplasmic Reticulum to Build Replication Neo-Organelles. mBio 2018, 9, e01253-18. [Google Scholar] [CrossRef]
- Morosky, S.; Lennemann, N.J.; Coyne, C.B. BPIFB6 Regulates Secretory Pathway Trafficking and Enterovirus Replication. J. Virol 2016, 90, 5098–5107. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Morosky, S.; Bomberger, J.; Stolz, D.B.; Jackson, W.T.; Coyne, C.B. BPIFB3 regulates autophagy and coxsackievirus B replication through a noncanonical pathway independent of the core initiation machinery. mBio 2014, 5, e02147. [Google Scholar] [CrossRef] [PubMed]
- Ao, D.; Guo, H.C.; Sun, S.Q.; Sun, D.H.; Fung, T.S.; Wei, Y.Q.; Han, S.C.; Yao, X.P.; Cao, S.Z.; Liu, D.X.; et al. Viroporin Activity of the Foot-and-Mouth Disease Virus Non-Structural 2B Protein. PLoS ONE 2015, 10, e0125828. [Google Scholar] [CrossRef] [PubMed]
- Ranjitha, H.B.; Ammanathan, V.; Guleria, N.; Hosamani, M.; Sreenivasa, B.P.; Dhanesh, V.V.; Santhoshkumar, R.; Sagar, B.K.C.; Mishra, B.P.; Singh, R.K.; et al. Foot-and-mouth disease virus induces PERK-mediated autophagy to suppress the antiviral interferon response. J. Cell Sci. 2020, 134, 240622. [Google Scholar] [CrossRef]
- Wessels, E.; Duijsings, D.; Lanke, K.H.; Melchers, W.J.; Jackson, C.L.; van Kuppeveld, F.J. Molecular determinants of the interaction between coxsackievirus protein 3A and guanine nucleotide exchange factor GBF1. J. Virol. 2007, 81, 5238–5245. [Google Scholar] [CrossRef]
- Yuan, L.; Kenny, S.J.; Hemmati, J.; Xu, K.; Schekman, R. TANGO1 and SEC12 are copackaged with procollagen I to facilitate the generation of large COPII carriers. Proc. Natl. Acad. Sci. USA 2018, 115, E12255–E12264. [Google Scholar] [CrossRef]
- Aridor, M. COPII gets in shape: Lessons derived from morphological aspects of early secretion. Traffic 2018, 19, 823–839. [Google Scholar] [CrossRef]
- Petrosyan, A.; Cheng, P.W.; Clemens, D.L.; Casey, C.A. Downregulation of the small GTPase SAR1A: A key event underlying alcohol-induced Golgi fragmentation in hepatocytes. Sci. Rep. 2015, 5, 17127. [Google Scholar] [CrossRef]
- Giraudo, C.G.; Maccioni, H.J. Endoplasmic reticulum export of glycosyltransferases depends on interaction of a cytoplasmic dibasic motif with Sar1. Mol. Biol. Cell 2003, 14, 3753–3766. [Google Scholar] [CrossRef]
- Quintero, C.A.; Giraudo, C.G.; Villarreal, M.; Montich, G.; Maccioni, H.J. Identification of a site in Sar1 involved in the interaction with the cytoplasmic tail of glycolipid glycosyltransferases. J. Biol. Chem. 2010, 285, 30340–30346. [Google Scholar] [CrossRef]
- de Castro Martin, I.F.; Fournier, G.; Sachse, M.; Pizarro-Cerda, J.; Risco, C.; Naffakh, N. Influenza virus genome reaches the plasma membrane via a modified endoplasmic reticulum and Rab11-dependent vesicles. Nat. Commun. 2017, 8, 1396. [Google Scholar] [CrossRef] [PubMed]
- Strating, J.R.; van Kuppeveld, F.J. Viral rewiring of cellular lipid metabolism to create membranous replication compartments. Curr. Opin. Cell Biol. 2017, 47, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Randall, G. (+) RNA virus replication com.mpartments: A safe home for (most) viral replication. Curr. Opin. Microbiol. 2016, 32, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Fernandez de Castro, I.; Tenorio, R.; Ortega-Gonzalez, P.; Knowlton, J.J.; Zamora, P.F.; Lee, C.H.; Fernandez, J.J.; Dermody, T.S.; Risco, C. A modified lysosomal organelle mediates nonlytic egress of reovirus. J. Cell Biol. 2020, 219, e201910131. [Google Scholar] [CrossRef]
- Belov, G.A.; Sztul, E. Rewiring of cellular membrane homeostasis by picornaviruses. J. Virol. 2014, 88, 9478–9489. [Google Scholar] [CrossRef]
- Melville, D.; Gorur, A.; Schekman, R. Fatty-acid binding protein 5 modulates the SAR1 GTPase cycle and enhances budding of large COPII cargoes. Mol. Biol. Cell 2019, 30, 387–399. [Google Scholar] [CrossRef]
- Long, K.R.; Yamamoto, Y.; Baker, A.L.; Watkins, S.C.; Coyne, C.B.; Conway, J.F.; Aridor, M. Sar1 assembly regulates membrane constriction and ER export. J. Cell Biol. 2010, 190, 115–128. [Google Scholar] [CrossRef]
- Wolk, B.; Buchele, B.; Moradpour, D.; Rice, C.M. A dynamic view of hepatitis C virus replication complexes. J. Virol. 2008, 82, 10519–10531. [Google Scholar] [CrossRef]
- Ci, Y.; Liu, Z.Y.; Zhang, N.N.; Niu, Y.; Yang, Y.; Xu, C.; Yang, W.; Qin, C.F.; Shi, L. Zika NS1-induced ER remodeling is essential for viral replication. J. Cell Biol. 2020, 219, e201903062. [Google Scholar] [CrossRef]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-W.; Jiang, Y.-F.; Chang, H.-W.; Cheng, I.-C. Foot-and-Mouth Disease Virus 3A Hijacks Sar1 and Sec12 for ER Remodeling in a COPII-Independent Manner. Viruses 2022, 14, 839. https://doi.org/10.3390/v14040839
Lee H-W, Jiang Y-F, Chang H-W, Cheng I-C. Foot-and-Mouth Disease Virus 3A Hijacks Sar1 and Sec12 for ER Remodeling in a COPII-Independent Manner. Viruses. 2022; 14(4):839. https://doi.org/10.3390/v14040839
Chicago/Turabian StyleLee, Heng-Wei, Yi-Fan Jiang, Hui-Wen Chang, and Ivan-Chen Cheng. 2022. "Foot-and-Mouth Disease Virus 3A Hijacks Sar1 and Sec12 for ER Remodeling in a COPII-Independent Manner" Viruses 14, no. 4: 839. https://doi.org/10.3390/v14040839
APA StyleLee, H. -W., Jiang, Y. -F., Chang, H. -W., & Cheng, I. -C. (2022). Foot-and-Mouth Disease Virus 3A Hijacks Sar1 and Sec12 for ER Remodeling in a COPII-Independent Manner. Viruses, 14(4), 839. https://doi.org/10.3390/v14040839