
Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity



Abstract
:1. Quasispecies and Lethal Mutagenesis
2. Mutation Rates and Fidelity of Viral Polymerases
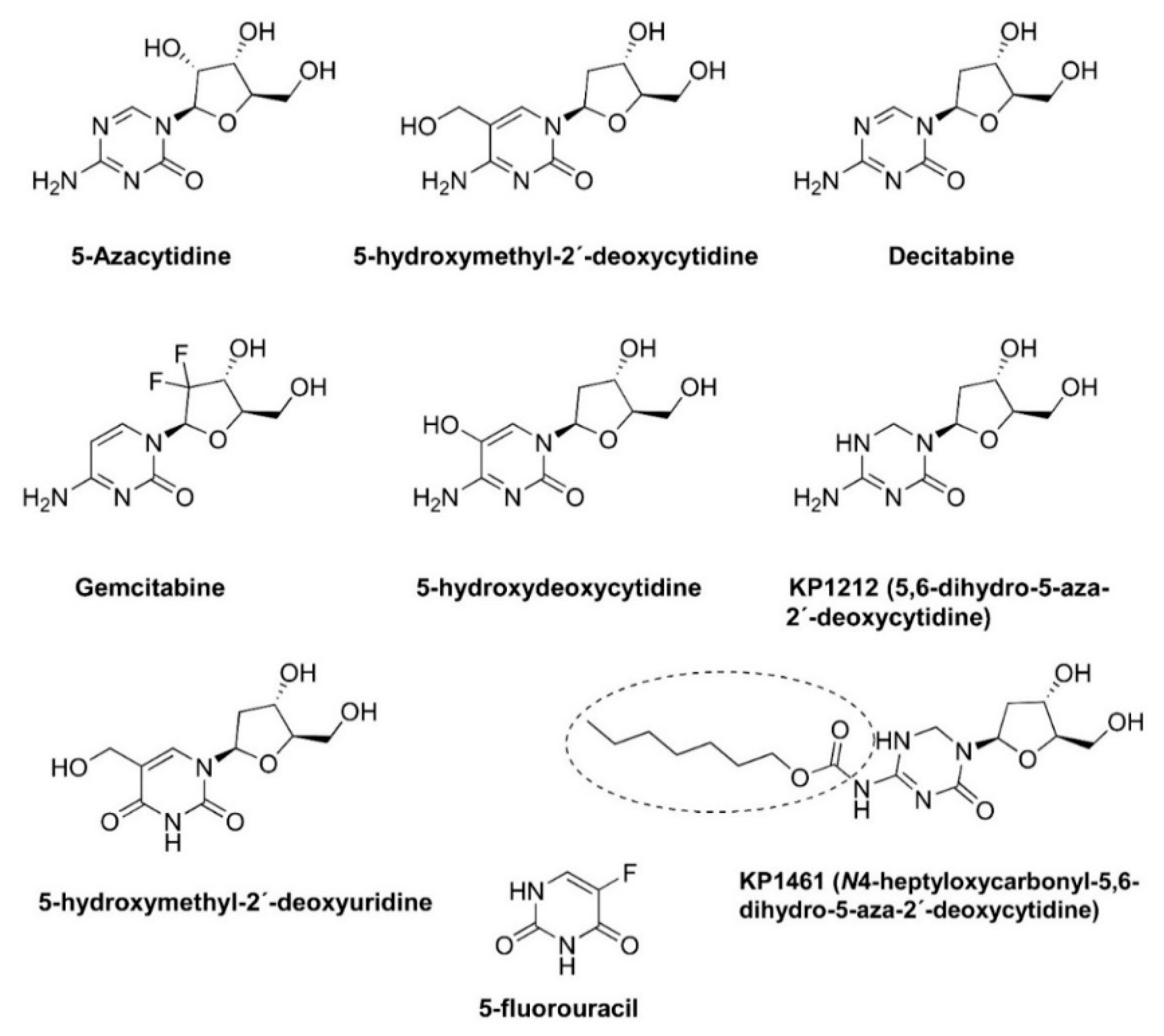
3. Driving HIV into Error Catastrophe and Preliminary Clinical Development of KP1212/KP1461 as an Antiretroviral Agent Causing Lethal Mutagenesis

{kind=link}
{kind=link}
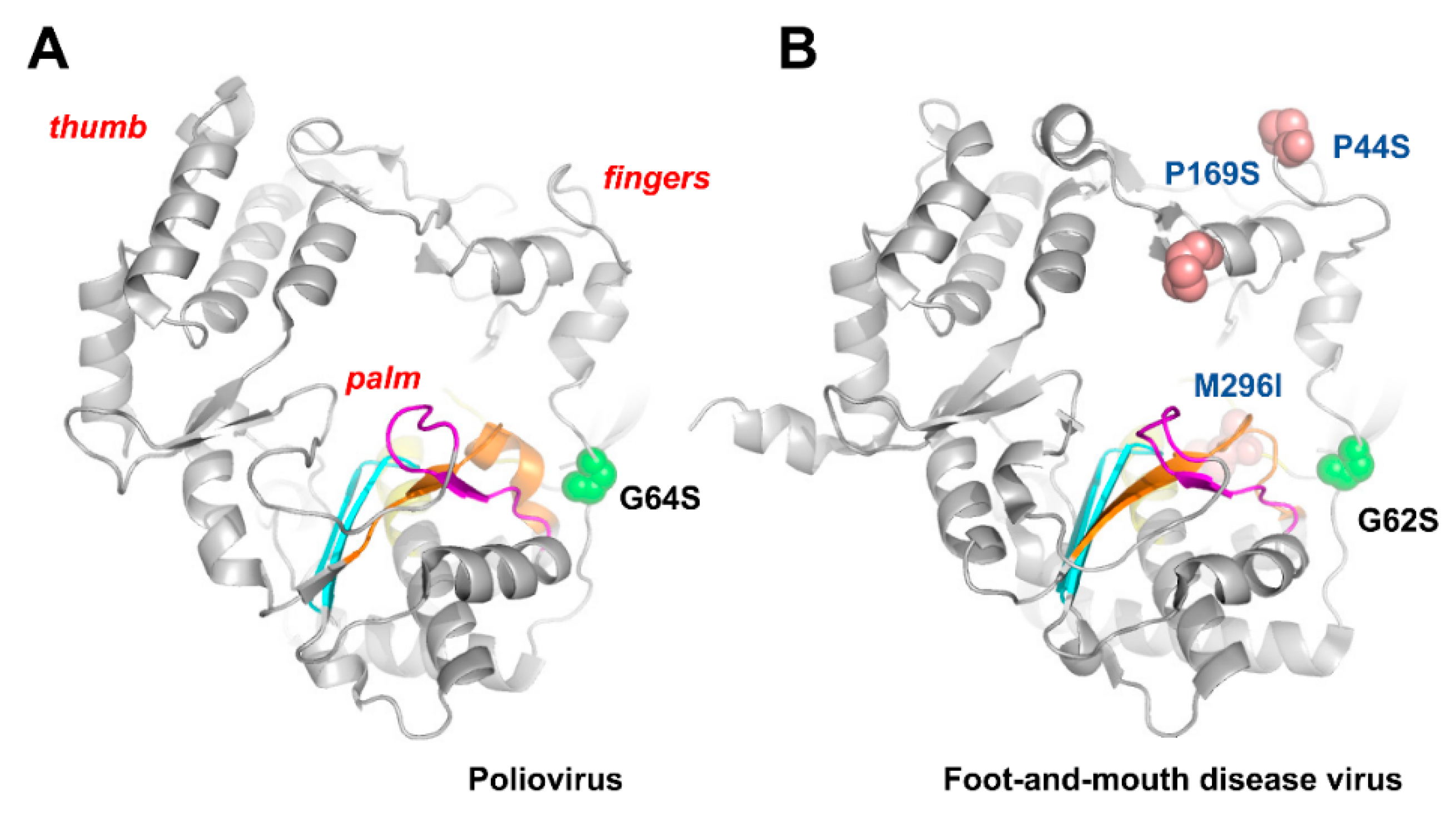{kind=link}
{kind=link}
| Mutagenic Nucleoside | Increase in Mutation Frequency | Mutational Preference | References |
|---|---|---|---|
| 5-azacytidine | 2.3-fold | G/C transversions | [45,47] |
| 5-fluorouracil | <1.5-fold | A/G, U/C transitions | [48] |
| 5-hydroxymethyl-2′-deoxycytidine | 3.4-fold | G→A, G→T | [49] |
| 5-hydroxymethyl-2′-deoxyuridine | 3.1-fold | A→G, G→C | [49] |
| Decitabine (5-aza-2′-deoxycytidine) | 4.1-fold | G/C transversions | [50] |
| Gemcitabine (2,2(′)-difluoro-2(′)-deoxycytidine) | <1.5-fold | [48] |
4. Lethal Mutagenesis in Non-Retroviral RNA Viruses: An Overview of Studies Showing the Effects of Base and Nucleoside Analogs
| Virus Names | Family/Genus | Mutagenic Base and Nucleoside Analogs | Refs. | |
|---|---|---|---|---|
| Positive-strand RNA viruses | ||||
| - | Poliovirus | Picornaviridae/Enterovirus | Ribavirin, 5-nitrocytidine, 6-(β-d-ribofuranosyl)-3,4-dihydro-8H-pyrimido [4,5-c][1,2]oxazin-7-one, and N-6-substituted purine analogs (JA28 and JA30) | [69,70,71,72] |
| Coxsackievirus | Picornaviridae/Enterovirus | Ribavirin, and N-6-substituted purine analogs | [72,73] | |
| Encephalomyocarditis virus | Picornaviridae/Cardiovirus | 5-Fluorouracil | [74] | |
| Foot-and-mouth disease virus | Picornaviridae/Aphthovirus | 5-Fluorouracil, ribavirin, and favipiravir | [74,75,76,77,78,79] | |
| Murine norovirus | Caliciviridae/Norovirus | Favipiravir | [80] | |
| Dengue virus | Flaviviridae/Flavivirus | 3-Hydroxy-2-pyrazinecarboxamide (T-1105), and its ribose derivative (T-1106) | [81] | |
| Usutu virus | Flaviviridae/Flavivirus | 5-Fluorouracil and favipiravir, while ribavirin effects are less pronounced. | [82] | |
| West Nile virus | Flaviviridae/Flavivirus | Ribavirin and favipiravir | [83,84] | |
| Zika virus | Flaviviridae/Flavivirus | Ribavirin and favipiravir | [82] | |
| GB virus B | Flaviviridae/Hepacivirus | Ribavirin | [85] | |
| Hepatitis C virus | Flaviviridae/Hepacivirus | Ribavirin and favipiravir | [86,87,88,89,90,91,92] | |
| Hepatitis E virus | Hepeviridae/Orthohepevirus | Ribavirin | [93] | |
| Venezuelan equine encephalitis virus | Togaviridae/Alphavirus | β-d-N4-hydroxycytidine (molnupiravir) | [94] | |
| SARS-CoV-2 | Coronaviridae/Betacoronavirus | Favipiravir and β-d-N4-hydroxycytidine (molnupiravir) | [68,95,96] | |
| Tobacco mosaic virus | Virgaviridae/Tobamovirus | 5-Fluorouracil | [97] | |
| Negative-strand RNA viruses | ||||
| Influenza A virus | Orthomyxoviridae/ Alphainfluenzavirus | Ribavirin, 5-azacytidine, 5-fluorouracil, and β-d-N4-hydroxycytidine (molnupiravir) | [98,99] | |
| Vesicular stomatitis virus | Rhabdoviridae/Vesiculovirus | 5-Fluorouracil | [74,100] | |
| Hantaan virus | Hantaviridae/Orthohantaviridae | Ribavirin | [101,102] | |
| Rift Valley fever virus | Phenuiviridae/Phlebovirus | Favipiravir | [103] | |
| Lymphocytic choriomeningitis virus | Arenaviridae/Mammarenavirus | 5-Fluorouracil | [74,104] | |
| Ebola virus | Filoviridae/Ebolavirus | Favipiravir | [105] | |
| Marburg virus | Filoviridae/Marburgvirus | Favipiravir | [105] | |
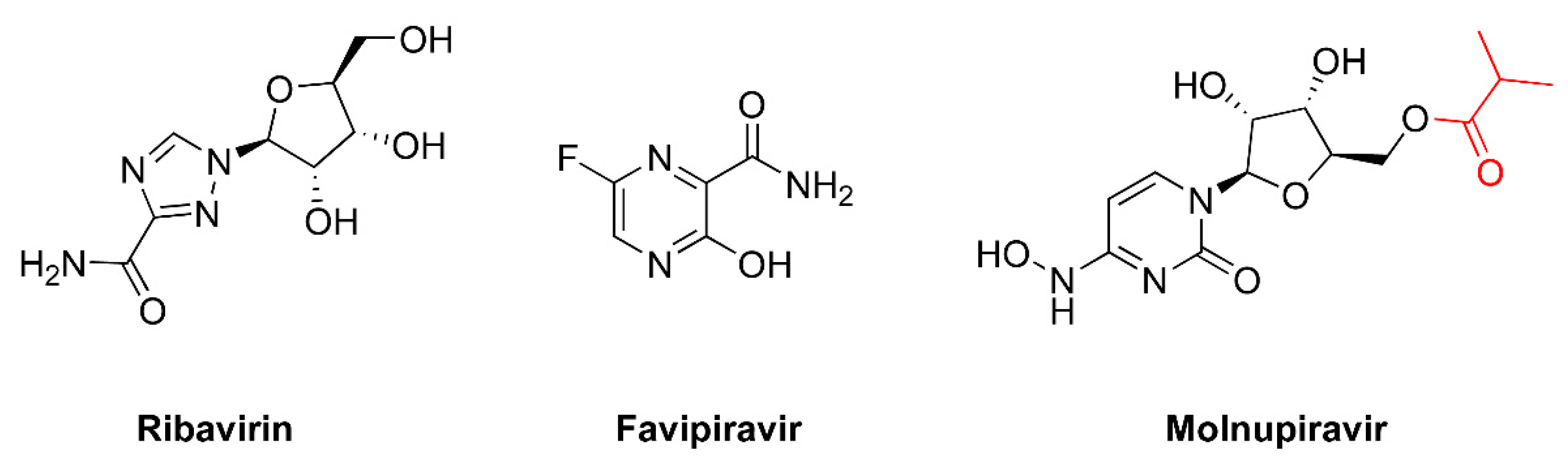
5. Mutagenic Effects of Ribavirin

6. Favipiravir as a Lethal Mutagenesis Agent
7. Molnupiravir as a Broad-Spectrum Antiviral Drug Effective against SARS-CoV-2

8. Future Directions and Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, E.; García-Crespo, C.; Perales, C. Historical perspective on the discovery of the quasispecies concept. Annu. Rev. Virol. 2021, 8, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. USA 2002, 99, 13374–13376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigen, M. From Strange Simplicity to Complex Familiarity: A Treatise on Matter, Information, Life and Thought; Oxford University Press: Cary, NC, USA, 2013. [Google Scholar]
- Weissmann, C.; Billeter, M.A.; Goodman, H.M.; Hindley, J.; Weber, H. Structure and function of phage RNA. Annu. Rev. Biochem. 1973, 42, 303–328. [Google Scholar] [CrossRef] [PubMed]
- Haruna, I.; Spiegelman, S. Recognition of size and sequence by an RNA replicase. Proc. Natl. Acad. Sci. USA 1965, 54, 1189–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haruna, I.; Spiegelman, S. Specific template requirments of RNA replicases. Proc. Natl. Acad. Sci. USA 1965, 54, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [Green Version]
- Perales, C.; Domingo, E. Antiviral strategies based on lethal mutagenesis and error threshold. Curr. Top. Microbiol. Immunol. 2016, 392, 323–339. [Google Scholar] [CrossRef]
- Holland, J.J.; Domingo, E.; de la Torre, J.C.; Steinhauer, D.A. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol. 1990, 64, 3960–3962. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.P.; Daifuku, R.; Loeb, L.A. Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 2004, 58, 183–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeb, L.A.; Essigmann, J.M.; Kazazi, F.; Zhang, J.; Rose, K.D.; Mullins, J.I. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. USA 1999, 96, 1492–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combe, M.; Sanjuán, R. Variation in RNA virus mutation rates across host cells. PLoS Pathog. 2014, 10, e1003855. [Google Scholar] [CrossRef] [Green Version]
- Peck, K.M.; Lauring, A.S. Complexities of viral mutation rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef] [Green Version]
- Coffin, J.M. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-Arias, L.; Sebastián-Martín, A.; Álvarez, M. Viral reverse transcriptases. Virus Res. 2017, 234, 153–176. [Google Scholar] [CrossRef]
- Menéndez-Arias, L. Mutation rates and intrinsic fidelity of retroviral reverse transcriptases. Viruses 2009, 1, 1137–1165. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, P.K.; Sun, G.; Yu, H.; Ron, Y.; Dougherty, J.P.; Preston, B.D. Mutational analysis of HIV-1 long terminal repeats to explore the relative contribution of reverse transcriptase and RNA polymerase II to viral mutagenesis. J. Biol. Chem. 2002, 277, 38053–38061. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-Arias, L. Molecular basis of fidelity of DNA synthesis and nucleotide specificity of retroviral reverse transcriptases. Prog. Nucleic Acid Res. Mol. Biol. 2002, 71, 91–147. [Google Scholar] [CrossRef]
- Elder, J.H.; Lerner, D.L.; Hasselkus-Light, C.S.; Fontenot, D.J.; Hunter, E.; Luciw, P.A.; Montelaro, R.C.; Phillips, T.R. Distinct subsets of retroviruses encode dUTPase. J. Virol. 1992, 66, 1791–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köppe, B.; Menéndez-Arias, L.; Oroszlan, S. Expression and purification of the mouse mammary tumor virus gag-pro transframe protein p30 and characterization of its dUTPase activity. J. Virol. 1994, 68, 2313–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, D.L.; Wagaman, P.C.; Phillips, T.R.; Prospero-García, O.; Henriksen, S.J.; Fox, H.S.; Bloom, F.E.; Elder, J.H. Increased mutation frequency of feline immunodeficiency virus lacking a functional deoxyuridine-triphosphatase. Proc. Natl. Acad. Sci. USA 1995, 92, 7480–7484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansky, L.M. The mutation rate of human immunodeficiency virus type 1 is influenced by the vpr gene. Virology 1996, 222, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Takaori-Kondo, A.; Shindo, K. HIV-1 Vif: A guardian of the virus that opens up a new era in the research field of restriction factors. Front Microbiol. 2013, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Shah, F.S.; Curr, K.A.; Hamburgh, M.E.; Parniak, M.; Mitsuya, H.; Arnez, J.G.; Prasad, V.R. Differential influence of nucleoside analog-resistance mutations K65R and L74V on the overall mutation rate and error specificity of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 2000, 275, 27037–27044. [Google Scholar] [CrossRef]
- Barrioluengo, V.; Alvarez, M.; Barbieri, D.; Menéndez-Arias, L. Thermostable HIV-1 group O reverse transcriptase variants with the same fidelity as murine leukaemia virus reverse transcriptase. Biochem. J. 2011, 436, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Martín-Hernández, A.M.; Domingo, E.; Menéndez-Arias, L. Human immunodeficiency virus type 1 reverse transcriptase: Role of Tyr115 in deoxynucleotide binding and misinsertion fidelity of DNA synthesis. EMBO J. 1996, 15, 4434–4442. [Google Scholar] [CrossRef]
- Cases-González, C.E.; Gutiérrez-Rivas, M.; Menéndez-Arias, L. Coupling ribose selection to fidelity of DNA synthesis: The role of Tyr-115 of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 2000, 275, 19759–19767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansky, L.M.; Le Rouzic, E.; Benichou, S.; Gajary, L.C. Influence of reverse transcriptase variants, drugs, and Vpr on human immunodeficiency virus type 1 mutant frequencies. J. Virol. 2003, 77, 2071–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peersen, O.B. Picornaviral polymerase structure, function, and fidelity modulation. Virus Res. 2017, 234, 4–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Gong, P. A structural view of the RNA-dependent RNA polymerases from the Flavivirus genus. Virus Res. 2017, 234, 34–43. [Google Scholar] [CrossRef]
- Hillen, H.S. Structure and function of SARS-CoV-2 polymerase. Curr. Opin. Virol. 2021, 48, 82–90. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.-L. Coronavirus RNA proofreading: Molecular basis and therapeutic targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef] [Green Version]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of SARS-CoV nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010, 6, e1000896. [Google Scholar] [CrossRef] [Green Version]
- Pathak, V.K.; Temin, H.M. 5-Azacytidine and RNA secondary structure increase the retrovirus mutation rate. J. Virol. 1992, 66, 3093–3100. [Google Scholar] [CrossRef] [Green Version]
- LaCasse, R.A.; Remington, K.M.; North, T.W. The mutation frequency of feline immunodeficiency virus enhanced by 3′-azido-3′-deoxythymidine. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 12, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Julias, J.G.; Kim, T.; Arnold, G.; Pathak, V.K. The antiretrovirus drug 3′-azido-3′-deoxythymidine increases the retrovirus mutation rate. J. Virol. 1997, 71, 4254–4263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansky, L.M.; Bernard, L.C. 3′-Azido-3′-deoxythymidine (AZT) and AZT-resistant reverse transcriptase can increase the in vivo mutation rate of human immunodeficiency virus type 1. J. Virol. 2000, 74, 9532–9539. [Google Scholar] [CrossRef] [Green Version]
- Dapp, M.J.; Clouser, C.L.; Patterson, S.; Mansky, L.M. 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J. Virol. 2009, 83, 11950–11958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, M.; McDaniel, Y.Z.; Daly, M.B.; Talledge, N.; Greggs, W.M., 3rd; Patterson, S.E.; Kim, B.; Mansky, L.M. Distinct antiretroviral mechanisms elicited by a viral mutagen. J. Mol. Biol. 2021, 433, 167111. [Google Scholar] [CrossRef] [PubMed]
- Rawson, J.M.O.; Daly, M.B.; Xie, J.; Clouser, C.L.; Landman, S.R.; Reilly, C.S.; Bonnac, L.; Kim, B.; Patterson, S.E.; Mansky, L.M. 5-Azacytidine enhances the mutagenesis of HIV-1 by reduction to 5-aza-2′-deoxycytidine. Antimicrob. Agents Chemother. 2016, 60, 2318–2325. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Jiménez, C.; Olivares, I.; de Ávila Lucas, A.I.; Toledano, V.; Gutiérrez-Rivas, M.; Lorenzo-Redondo, R.; Grande-Pérez, A.; Domingo, E.; López-Galíndez, C. Mutagen-mediated enhancement of HIV-1 replication in persistently infected cells. Virology 2012, 424, 147–153. [Google Scholar] [CrossRef]
- Vivet-Boudou, V.; Isel, C.; El Safadi, Y.; Smyth, R.P.; Laumond, G.; Moog, C.; Paillart, J.-C.; Marquet, R. Evaluation of anti-HIV-1 mutagenic nucleoside analogues. J. Biol. Chem. 2015, 290, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Rawson, J.M.O.; Landman, S.R.; Reilly, C.S.; Bonnac, L.; Patterson, S.E.; Mansky, L.M. Lack of mutational hot spots during decitabine-mediated HIV-1 mutagenesis. Antimicrob. Agents Chemother. 2015, 59, 6834–6843. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-Arias, L. Targeting HIV: Antiretroviral therapy and development of drug resistance. Trends Pharmacol. Sci. 2002, 23, 381–388. [Google Scholar] [CrossRef]
- Menéndez-Arias, L.; Delgado, R. Update and latest advances in antiretroviral therapy. Trends Pharmacol. Sci. 2022, 43, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Tapia, N.; Fernàndez, G.; Parera, M.; Gómez-Mariano, G.; Clotet, B.; Quiñones-Mateu, M.; Domingo, E.; Martínez, M.A. Combination of a mutagenic agent with a reverse transcriptase inhibitor results in systematic inhibition of HIV-1 infection. Virology 2005, 338, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seier, T.; Zilberberg, G.; Zeiger, D.M.; Lovett, S.T. Azidothymidine and other chain terminators are mutagenic for template-switch-generated genetic mutations. Proc. Natl. Acad. Sci. USA 2012, 109, 6171–6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, K.S.; Brabant, W.; Styrchak, S.; Gall, A.; Daifuku, R. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antivir. Res. 2005, 67, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Fedeles, B.I.; Singh, V.; Peng, C.S.; Silvestre, K.J.; Simi, A.K.; Simpson, J.H.; Tokmakoff, A.; Essigmann, J.M. Tautomerism provides a molecular explanation for the mutagenic properties of the anti-HIV nucleoside 5-aza-5,6-dihydro-2′-deoxycytidine. Proc. Natl. Acad. Sci. USA 2014, 111, E3252–E3259. [Google Scholar] [CrossRef] [Green Version]
- Mullins, J.I.; Heath, L.; Hughes, J.P.; Kicha, J.; Styrchak, S.; Wong, K.G.; Rao, U.; Hansen, A.; Harris, K.S.; Laurent, J.-P.; et al. Mutation of HIV-1 genomes in a clinical population treated with the mutagenic nucleoside KP1461. PLoS ONE 2011, 6, e15135. [Google Scholar] [CrossRef] [Green Version]
- Fontecave, M.; Lepoivre, M.; Elleingand, E.; Gerez, C.; Guittet, O. Resveratrol, a remarkable inhibitor of ribonucleotide reductase. FEBS Lett. 1998, 421, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Rawson, J.M.; Heineman, R.H.; Beach, L.B.; Martin, J.L.; Schnettler, E.K.; Dapp, M.J.; Patterson, S.E.; Mansky, L.M. 5,6-Dihydro-5-aza-2′-deoxycytidine potentiates the anti-HIV-1 activity of ribonucleotide reductase inhibitors. Bioorg. Med. Chem. 2013, 21, 7222–7228. [Google Scholar] [CrossRef] [Green Version]
- Guarino, E.; Salguero, I.; Kearsey, S.E. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin. Cell Dev. Biol. 2014, 30, 97–103. [Google Scholar] [CrossRef]
- Musiałek, M.W.; Rybaczek, D. Hydroxyurea-The good, the bad and the ugly. Genes 2021, 12, 1096. [Google Scholar] [CrossRef]
- Clouser, C.L.; Patterson, S.E.; Mansky, L.M. Exploiting drug repositioning for discovery of a novel HIV combination therapy. J. Virol. 2010, 84, 9301–9309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawson, J.M.O.; Roth, M.E.; Xie, J.; Daly, M.B.; Clouser, C.L.; Landman, S.R.; Reilly, C.S.; Bonnac, L.; Kim, B.; Patterson, S.E.; et al. Synergistic reduction of HIV-1 infectivity by 5-azacytidine and inhibitors of ribonucleotide reductase. Bioorg. Med. Chem. 2016, 24, 2410–2422. [Google Scholar] [CrossRef] [PubMed]
- Beach, L.B.; Rawson, J.M.; Kim, B.; Patterson, S.E.; Mansky, L.M. Novel inhibitors of human immunodeficiency virus type 2 infectivity. J. Gen. Virol. 2014, 95 Pt 12, 2778–2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, M.; Sebastián-Martín, A.; García-Marquina, G.; Menéndez-Arias, L. Fidelity of classwide-resistant HIV-2 reverse transcriptase and differential contribution of K65R to the accuracy of HIV-1 and HIV-2 reverse transcriptases. Sci. Rep. 2017, 7, 44834. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, Y.Z.; Patterson, S.E.; Mansky, L.M. Distinct dual antiviral mechanism that enhances hepatitis B virus mutagenesis and reduces viral DNA synthesis. Antiviral. Res. 2019, 170, 104540. [Google Scholar] [CrossRef]
- Holland, J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; VandePol, S. Rapid evolution of RNA genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef]
- Zhou, S.; Hill, C.S.; Sarkar, S.; Tse, L.V.; Woodburn, B.M.D.; Schinazi, R.F.; Sheahan, T.P.; Baric, R.S.; Heise, M.T.; Swanstrom, R. β-D-N4-hydroxycytidine inhibits SARS-CoV-2 through lethal mutagenesis but is also mutagenic to mammalian cells. J. Infect. Dis. 2021, 224, 415–419. [Google Scholar] [CrossRef]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [CrossRef]
- Harki, D.A.; Graci, J.D.; Galarraga, J.E.; Chain, W.J.; Cameron, C.E.; Peterson, B.R. Synthesis and antiviral activity of 5-substituted cytidine analogues: Identification of a potent inhibitor of viral RNA-dependent RNA polymerases. J. Med. Chem. 2006, 49, 6166–6169. [Google Scholar] [CrossRef] [Green Version]
- Graci, J.D.; Harki, D.A.; Korneeva, V.S.; Edathil, J.P.; Too, K.; Franco, D.; Smidansky, E.D.; Paul, A.V.; Peterson, B.R.; Brown, D.M.; et al. Lethal mutagenesis of poliovirus mediated by a mutagenic pyrimidine analogue. J. Virol. 2007, 81, 11256–11266. [Google Scholar] [CrossRef] [Green Version]
- Graci, J.D.; Too, K.; Smidansky, E.D.; Edathil, J.P.; Barr, E.W.; Harki, D.A.; Galarraga, J.E.; Bollinger, J.M., Jr.; Peterson, B.R.; Loakes, D.; et al. Lethal mutagenesis of picornaviruses with N-6-modified purine nucleoside analogues. Antimicrob. Agents Chemother. 2008, 52, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graci, J.D.; Gnädig, N.F.; Galarraga, J.E.; Castro, C.; Vignuzzi, M.; Cameron, C.E. Mutational robustness of an RNA virus influences sensitivity to lethal mutagenesis. J. Virol. 2012, 86, 2869–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, H.; Tejero, H.; de la Torre, J.C.; Domingo, E.; Martín, V. Mutagenesis-mediated virus extinction: Virus-dependent effect of viral load on sensitivity to lethal defection. PLoS ONE 2012, 7, e32550. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, A.; Pariente, N.; Menéndez-Arias, L.; Domingo, E. Curing of foot-and-mouth disease virus from persistently infected cells by ribavirin involves enhanced mutagenesis. Virology 2003, 311, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Pariente, N.; Airaksinen, A.; Domingo, E. Mutagenesis versus inhibition in the efficiency of extinction of foot-and-mouth disease virus. J. Virol. 2003, 77, 7131–7138. [Google Scholar] [CrossRef] [Green Version]
- Sierra, M.; Airaksinen, A.; González-López, C.; Agudo, R.; Arias, A.; Domingo, E. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: Implications for error catastrophe. J. Virol. 2007, 81, 2012–2024. [Google Scholar] [CrossRef] [Green Version]
- Agudo, R.; Ferrer-Orta, C.; Arias, A.; de la Higuera, I.; Perales, C.; Pérez-Luque, R.; Verdaguer, N.; Domingo, E. A multi-step process of viral adaptation to a mutagenic nucleoside analogue by modulation of transition types leads to extinction-escape. PLoS Pathog. 2010, 6, e1001072. [Google Scholar] [CrossRef] [Green Version]
- de Avila, A.I.; Moreno, E.; Perales, C.; Domingo, E. Favipiravir can evoke lethal mutagenesis and extinction of foot-and-mouth disease virus. Virus Res. 2017, 233, 105–112. [Google Scholar] [CrossRef]
- Arias, A.; Thorne, L.; Goodfellow, I. Favipiravir elicits antiviral mutagenesis during virus replication in vivo. Elife 2014, 3, e03679. [Google Scholar] [CrossRef]
- Qiu, L.; Patterson, S.E.; Bonnac, L.F.; Geraghty, R.J. Nucleobases and corresponding nucleosides display potent antiviral activities against dengue virus possibly through viral lethal mutagenesis. PLoS Negl. Trop. Dis. 2018, 12, e0006421. [Google Scholar] [CrossRef]
- Bassi, M.R.; Sempere, R.N.; Meyn, P.; Polacek, C.; Arias, A. Extinction of Zika virus and Usutu virus by lethal mutagenesis reveals different patterns of sensitivity to three mutagenic drugs. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.W.; Smee, D.F.; Julander, J.G.; Yamshchikov, V.F.; Sidwell, R.W.; Morrey, J.D. Error-prone replication of West Nile virus caused by ribavirin. Antiviral. Res. 2005, 67, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Romero, E.; Jiménez de Oya, N.; Domingo, E.; Saiz, J.C. Extinction of West Nile virus by favipiravir through lethal mutagenesis. Antimicrob. Agents Chemother. 2017, 61, e01400-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanford, R.E.; Chavez, D.; Guerra, B.; Lau, J.Y.; Hong, Z.; Brasky, K.M.; Beames, B. Ribavirin induces error-prone replication of GB virus B in primary tamarin hepatocytes. J. Virol. 2001, 75, 8074–8081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, A.M.; Hiasa, Y.; He, W.; Terella, A.; Schmidt, E.V.; Chung, R.T. Viral RNA mutations are region specific and increased by ribavirin in a full-length hepatitis C virus replication system. J. Virol. 2002, 76, 8505–8517. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Liu, R.; Baroudy, B.M.; Malcolm, B.A.; Reyes, G.R. The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology 2003, 310, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, J.M.; González-Candelas, F.; Moya, A.; Sanjuán, R. Effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. J. Virol. 2009, 83, 5760–5764. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Prieto, A.M.; Sheldon, J.; Grande-Pérez, A.; Tejero, H.; Gregori, J.; Quer, J.; Esteban, J.I.; Domingo, E.; Perales, C. Extinction of hepatitis C virus by ribavirin in hepatoma cells involves lethal mutagenesis. PLoS ONE 2013, 8, e71039. [Google Scholar] [CrossRef] [Green Version]
- Dietz, J.; Schelhorn, S.-E.; Fitting, D.; Mihm, U.; Susser, S.; Welker, M.-W.; Füller, C.; Däumer, M.; Teuber, G.; Wedemeyer, H.; et al. Deep sequencing reveals mutagenic effects of ribavirin during monotherapy of hepatitis C virus genotype 1-infected patients. J. Virol. 2013, 87, 6172–6181. [Google Scholar] [CrossRef] [Green Version]
- Gallego, I.; Soria, M.E.; Gregori, J.; de Ávila, A.I.; García-Crespo, C.; Moreno, E.; Gadea, I.; Esteban, J.; Fernández-Roblas, R.; Esteban, J.I.; et al. Synergistic lethal mutagenesis of hepatitis C virus. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- de Ávila, A.I.; Gallego, I.; Soria, M.E.; Gregori, J.; Quer, J.; Esteban, J.I.; Rice, C.M.; Domingo, E.; Perales, C. Lethal mutagenesis of hepatitis C virus induced by favipiravir. PLoS ONE 2016, 11, e0164691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todt, D.; Walter, S.; Brown, R.; Steinmann, E. Mutagenic effects of ribavirin on hepatitis E virus—Viral extinction versus selection of fitness-enhancing mutations. Viruses 2016, 8, 283. [Google Scholar] [CrossRef] [Green Version]
- Urakova, N.; Kuznetsova, V.; Crossman, D.K.; Sokratian, A.; Guthrie, D.B.; Kolykhalov, A.A.; Lockwood, M.A.; Natchus, M.G.; Crowley, M.R.; Painter, G.R.; et al. β-D-N4-hydroxycytidine is a potent anti-alphavirus compound that induces a high level of mutations in the viral genome. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, A.; Selisko, B.; Le, N.-T.-T.; Huchting, J.; Touret, F.; Piorkowski, G.; Fattorini, V.; Ferron, F.; Decroly, E.; Meier, C.; et al. Rapid incorporation of favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat. Commun. 2020, 11, 4682. [Google Scholar] [CrossRef]
- Agostini, M.L.; Pruijssers, A.J.; Chappell, J.D.; Gribble, J.; Lu, X.; Andres, E.L.; Bluemling, G.R.; Lockwood, M.A.; Sheahan, T.P.; Sims, A.C.; et al. Small-molecule antiviral β-D-N4-hydroxycytidine inhibits a proofreading-intact coronavirus with a high genetic barrier to resistance. J. Virol. 2019, 93, e01348-19. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Martínez, L.; Brichette-Mieg, I.; Pineño-Ramos, A.; Domínguez-Huerta, G.; Grande-Pérez, A. Lethal mutagenesis of an RNA plant virus via lethal defection. Sci. Rep. 2018, 8, 1444. [Google Scholar] [CrossRef] [Green Version]
- Pauly, M.D.; Lauring, A.S. Effective lethal mutagenesis of influenza virus by three nucleoside analogs. J. Virol. 2015, 89, 3584–3597. [Google Scholar] [CrossRef] [Green Version]
- Toots, M.; Yoon, J.-J.; Cox, R.M.; Hart, M.; Sticher, Z.M.; Makhsous, N.; Plesker, R.; Barrena, A.H.; Reddy, P.G.; Mitchell, D.G.; et al. Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia. Sci. Transl. Med. 2019, 11, eaax5866. [Google Scholar] [CrossRef]
- Grande-Pérez, A.; Lázaro, E.; Lowenstein, P.; Domingo, E.; Manrubia, S.C. Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. USA 2005, 102, 4448–4452. [Google Scholar] [CrossRef] [Green Version]
- Severson, W.E.; Schmaljohn, C.S.; Javadian, A.; Jonsson, C.B. Ribavirin causes error catastrophe during Hantaan virus replication. J. Virol. 2003, 77, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Chung, D.-H.; Sun, Y.; Parker, W.B.; Arterburn, J.B.; Bartolucci, A.; Jonsson, C.B. Ribavirin reveals a lethal threshold of allowable mutation frequency for Hantaan virus. J. Virol. 2007, 81, 11722–11729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrego, B.; de Ávila, A.I.; Domingo, E.; Brun, A. Lethal mutagenesis of Rift Valley fever virus induced by favipiravir. Antimicrob. Agents Chemother. 2019, 63, e00669-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Jarabo, C.M.; Ly, C.; Domingo, E.; de la Torre, J.C. Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV). Virology 2003, 308, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Espy, N.; Nagle, E.; Pfeffer, B.; Garcia, K.; Chitty, A.J.; Wiley, M.; Sanchez-Lockhart, M.; Bavari, S.; Warren, T.; Palacios, G. T-705 induces lethal mutagenesis in Ebola and Marburg populations in macaques. Antiviral. Res. 2019, 170, 104529. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Andino, R. Ribavirin and lethal mutagenesis of poliovirus: Molecular mechanisms, resistance and biological implications. Virus Res. 2005, 107, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Olivencia, G.; Estébanez, M.; Membrillo, F.J.; Ybarra, M.D.C. Uso de ribavirina en virus distintos de la hepatitis C. una revisión de la evidencia. Enferm. Infecc. Microbiol. Clin. (Engl.) 2019, 37, 602–608. [Google Scholar] [CrossRef]
- Graci, J.D.; Cameron, C.E. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 2006, 16, 37–48. [Google Scholar] [CrossRef]
- Paeshuyse, J.; Dallmeier, K.; Neyts, J. Ribavirin for the treatment of chronic hepatitis C virus infection: A review of the proposed mechanisms of action. Curr. Opin. Virol. 2011, 1, 590–598. [Google Scholar] [CrossRef]
- Kentsis, A.; Topisirovic, I.; Culjkovic, B.; Shao, L.; Borden, K.L.B. Ribavirin suppresses EIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc. Natl. Acad. Sci. USA 2004, 101, 18105–18110. [Google Scholar] [CrossRef] [Green Version]
- Sidwell, R.W.; Huffman, J.H.; Khare Lois, G.P.; Allen, B.; Witkowski Roland, J.T.; Robins, K. Broad-spectrum antiviral activity of virazole: 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 1972, 177, 705–706. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Aliota, M.T.; Bonnac, L.F. Broad-spectrum antiviral strategies and nucleoside analogues. Viruses 2021, 13, 667. [Google Scholar] [CrossRef]
- Arnold, J.J.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3Dpol) is sufficient for template switching in vitro. J. Biol. Chem. 1999, 274, 2706–2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohara, D.W.; Crotty, S.; Arnold, J.J.; Yoder, J.D.; Andino, R.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3Dpol): Structural, biochemical, and biological analysis of conserved structural motifs a and b. J. Biol. Chem. 2000, 275, 25523–25532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Orta, C.; Sierra, M.; Agudo, R.; de la Higuera, I.; Arias, A.; Pérez-Luque, R.; Escarmís, C.; Domingo, E.; Verdaguer, N. Structure of foot-and-mouth disease virus mutant polymerases with reduced sensitivity to ribavirin. J. Virol. 2010, 84, 6188–6199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, K.-C.; Lindsay, K.L.; Lee, K.-J.; Liu, W.-C.; He, J.-W.; Milstein, S.L.; Lai, M.M.C. Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology 2003, 38, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Lutchman, G.; Danehower, S.; Song, B.-C.; Liang, T.J.; Hoofnagle, J.H.; Thomson, M.; Ghany, M.G. Mutation rate of the hepatitis C virus NS5B in patients undergoing treatment with ribavirin monotherapy. Gastroenterology 2007, 132, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Mejer, N.; Galli, A.; Ramirez, S.; Fahnøe, U.; Benfield, T.; Bukh, J. Ribavirin inhibition of cell-culture infectious hepatitis C genotype 1-3 viruses is strain-dependent. Virology 2020, 540, 132–140. [Google Scholar] [CrossRef]
- Mejer, N.; Fahnøe, U.; Galli, A.; Ramirez, S.; Weiland, O.; Benfield, T.; Bukh, J. Mutations identified in the hepatitis C virus (HCV) polymerase of patients with chronic HCV treated with ribavirin cause resistance and affect viral replication fidelity. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Gong, P.; Peersen, O.B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2010, 107, 22505–22510. [Google Scholar] [CrossRef] [Green Version]
- Gallego, I.; Gregori, J.; Soria, M.E.; García-Crespo, C.; García-Álvarez, M.; Gómez-González, A.; Valiergue, R.; Gómez, J.; Esteban, J.I.; Quer, J.; et al. Resistance of high fitness hepatitis C virus to lethal mutagenesis. Virology 2018, 523, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Delang, L.; Abdelnabi, R.; Neyts, J. Favipiravir as a potential countermeasure against neglected and emerging RNA viruses. Antiviral. Res. 2018, 153, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Jordan, P.C.; Stevens, S.K.; Deval, J. Nucleosides for the treatment of respiratory RNA virus infections. Antivir. Chem. Chemother. 2018, 26, 2040206618764483. [Google Scholar] [CrossRef] [PubMed]
- Sangawa, H.; Komeno, T.; Nishikawa, H.; Yoshida, A.; Takahashi, K.; Nomura, N.; Furuta, Y. Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob. Agents Chemother. 2013, 57, 5202–5208. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Smith, L.K.; Rajwanshi, V.K.; Kim, B.; Deval, J. The ambiguous base-pairing and high substrate efficiency of T-705 (favipiravir) ribofuranosyl 5’-triphosphate towards influenza A virus polymerase. PLoS ONE 2013, 8, e68347. [Google Scholar] [CrossRef]
- Baranovich, T.; Wong, S.-S.; Armstrong, J.; Marjuki, H.; Webby, R.J.; Webster, R.G.; Govorkova, E.A. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 2013, 87, 3741–3751. [Google Scholar] [CrossRef] [Green Version]
- Goldhill, D.H.; Te Velthuis, A.J.W.; Fletcher, R.A.; Langat, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA 2018, 115, 11613–11618. [Google Scholar] [CrossRef] [Green Version]
- Goldhill, D.H.; Yan, A.; Frise, R.; Zhou, J.; Shelley, J.; Gallego Cortés, A.; Miah, S.; Akinbami, O.; Galiano, M.; Zambon, M.; et al. Favipiravir-resistant influenza A virus shows potential for transmission. PLoS Pathog. 2021, 17, e1008937. [Google Scholar] [CrossRef]
- Delang, L.; Segura Guerrero, N.; Tas, A.; Quérat, G.; Pastorino, B.; Froeyen, M.; Dallmeier, K.; Jochmans, D.; Herdewijn, P.; Bello, F.; et al. Mutations in the Chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. J. Antimicrob. Chemother. 2014, 69, 2770–2784. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.; Yuan, S.; Gao, Q.; Lan, K.; Altmeyer, R.; Zou, G. In vitro assessment of combinations of enterovirus inhibitors against enterovirus 71. Antimicrob. Agents Chemother. 2016, 60, 5357–5367. [Google Scholar] [CrossRef] [Green Version]
- Naydenova, K.; Muir, K.W.; Wu, L.-F.; Zhang, Z.; Coscia, F.; Peet, M.J.; Castro-Hartmann, P.; Qian, P.; Sader, K.; Dent, K.; et al. Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc. Natl. Acad. Sci. USA 2021, 118, e2021946118. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Dandapat, J.; Saudagar, P.; Uversky, V.N.; Tripathi, T. Interface-based design of the favipiravir-binding site in SARS-CoV-2 RNA-dependent RNA polymerase reveals mutations conferring resistance to chain termination. FEBS Lett. 2021, 595, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Guo, S.; Yi, D.; Li, Q.; Ma, L.; Zhang, Y.; Wang, J.; Li, X.; Guo, F.; Lin, R.; et al. A cell-based assay to discover inhibitors of SARS-CoV-2 RNA dependent RNA polymerase. Antiviral. Res. 2021, 190, 105078. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H., 3rd; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Götte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef] [PubMed]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Jena, N.R. Role of different tautomers in the base-pairing abilities of some of the vital antiviral drugs used against COVID-19. Phys. Chem. Chem. Phys. 2020, 22, 28115–28122. [Google Scholar] [CrossRef]
- Rosenke, K.; Hansen, F.; Schwarz, B.; Feldmann, F.; Haddock, E.; Rosenke, R.; Barbian, K.; Meade-White, K.; Okumura, A.; Leventhal, S.; et al. Orally delivered MK-4482 inhibits SARS-CoV-2 replication in the Syrian hamster model. Nat. Commun. 2021, 12, 2295. [Google Scholar] [CrossRef]
- Cox, R.M.; Wolf, J.D.; Plemper, R.K. Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat. Microbiol. 2021, 6, 11–18. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; De los Reyes, V.; Martín-Quirós, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for oral treatment of COVID-19 in nonhospitalized patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef]
- Fischer, W.A., 2nd; Eron, J.J., Jr.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.J.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. A phase 2a clinical trial of molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci. Transl. Med. 2022, 14, eabl7430. [Google Scholar] [CrossRef] [PubMed]
- Abdelnabi, R.; Foo, C.S.; De Jonghe, S.; Maes, P.; Weynand, B.; Neyts, J. Molnupiravir inhibits replication of the emerging SARS-CoV-2 variants of concern in a hamster infection model. J. Infect. Dis. 2021, 224, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, Y.; Lavrijsen, M.; Lamers, M.M.; de Vries, A.C.; Rottier, R.J.; Bruno, M.J.; Peppelenbosch, M.P.; Haagmans, B.L.; Pan, Q. SARS-CoV-2 omicron variant is highly sensitive to molnupiravir, nirmatrelvir, and the combination. Cell Res. 2022, 32, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Vangeel, L.; Chiu, W.; De Jonghe, S.; Maes, P.; Slechten, B.; Raymenants, J.; André, E.; Leyssen, P.; Neyts, J.; Jochmans, D. Remdesivir, molnupiravir and nirmatrelvir remain active against SARS-CoV-2 omicron and other variants of concern. Antiviral. Res. 2022, 198, 105252. [Google Scholar] [CrossRef]
- Menéndez-Arias, L. Decoding molnupiravir-induced mutagenesis in SARS-CoV-2. J. Biol. Chem. 2021, 297, 100867. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Foo, C.S.; Kaptein, S.J.F.; Zhang, X.; Do, T.N.D.; Langendries, L.; Vangeel, L.; Breuer, J.; Pang, J.; Williams, R.; et al. The combined treatment of molnupiravir and favipiravir results in a potentiation of antiviral efficacy in a SARS-CoV-2 hamster infection model. EBioMedicine 2021, 72, 103595. [Google Scholar] [CrossRef]
- Schultz, D.C.; Johnson, R.M.; Ayyanathan, K.; Miller, J.; Whig, K.; Kamalia, B.; Dittmar, M.; Weston, S.; Hammond, H.L.; Dillen, C.; et al. Pyrimidine inhibitors synergize with nucleoside analogues to block SARS-CoV-2. Nature 2022, 604, 134–140. [Google Scholar] [CrossRef]
- Painter, G.R.; Natchus, M.G.; Cohen, O.; Holman, W.; Painter, W.P. Developing a direct acting, orally available antiviral agent in a pandemic: The evolution of molnupiravir as a potential treatment for COVID-19. Curr. Opin. Virol. 2021, 50, 17–22. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, T.; Xi, J.; Zhang, G.; Wang, T.; Liu, W.; You, X.; Zhang, X.; Xia, Z.; Luan, Y. PIG-A gene mutation as a genotoxicity biomarker in human population studies: An investigation in lead-exposed workers. Environ. Mol. Mutagen. 2020, 61, 611–621. [Google Scholar] [CrossRef]
- Waters, M.D.; Warren, S.; Hughes, C.; Lewis, P.; Zhang, F. Human genetic risk of treatment with antiviral nucleoside analog drugs that induce lethal mutagenesis: The special case of molnupiravir. Environ. Mol. Mutagen. 2022, 63, 37–63. [Google Scholar] [CrossRef]
- Ma, Y.; Frutos-Beltrán, E.; Kang, D.; Pannecouque, C.; De Clercq, E.; Menéndez-Arias, L.; Liu, X.; Zhan, P. Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chem. Soc. Rev. 2021, 50, 4514–4540. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadj Hassine, I.; Ben M’hadheb, M.; Menéndez-Arias, L. Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity. Viruses 2022, 14, 841. https://doi.org/10.3390/v14040841
Hadj Hassine I, Ben M’hadheb M, Menéndez-Arias L. Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity. Viruses. 2022; 14(4):841. https://doi.org/10.3390/v14040841
Chicago/Turabian StyleHadj Hassine, Ikbel, Manel Ben M’hadheb, and Luis Menéndez-Arias. 2022. "Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity" Viruses 14, no. 4: 841. https://doi.org/10.3390/v14040841
APA StyleHadj Hassine, I., Ben M’hadheb, M., & Menéndez-Arias, L. (2022). Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity. Viruses, 14(4), 841. https://doi.org/10.3390/v14040841