
ADRV 12L: A Ranaviral Putative Rad2 Family Protein Involved in DNA Recombination and Repair


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses and Cells
2.2. Sequence Analysis
2.3. Antibody Preparation
2.4. RT-PCR and Western Blot Analysis
2.5. Cytarabine Treatment
2.6. Immunofluorescence Microscopy
2.7. Generation of 12L Knockout Virus
2.8. Plaque Assay
2.9. One-Step Virus Growth Curves
2.10. Luc-HR Assay
2.11. DSBR Assay
3. Results
3.1. Sequence Characteristics of ADRV 12L
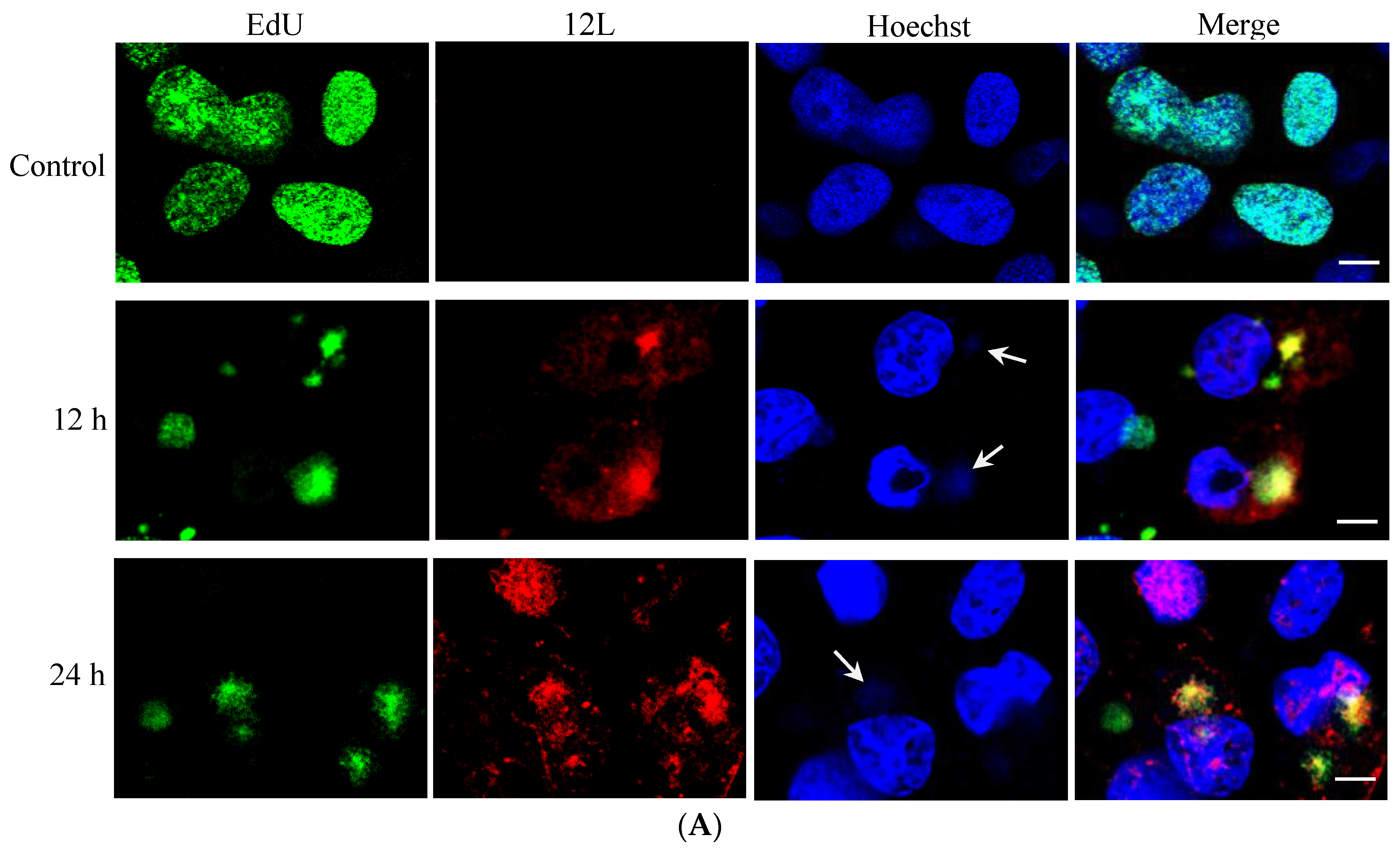
3.2. Temporal Expression Pattern and Subcellular Localization of ADRV 12L during Infection
3.3. Construction of 12L Deleted Recombinant Virus
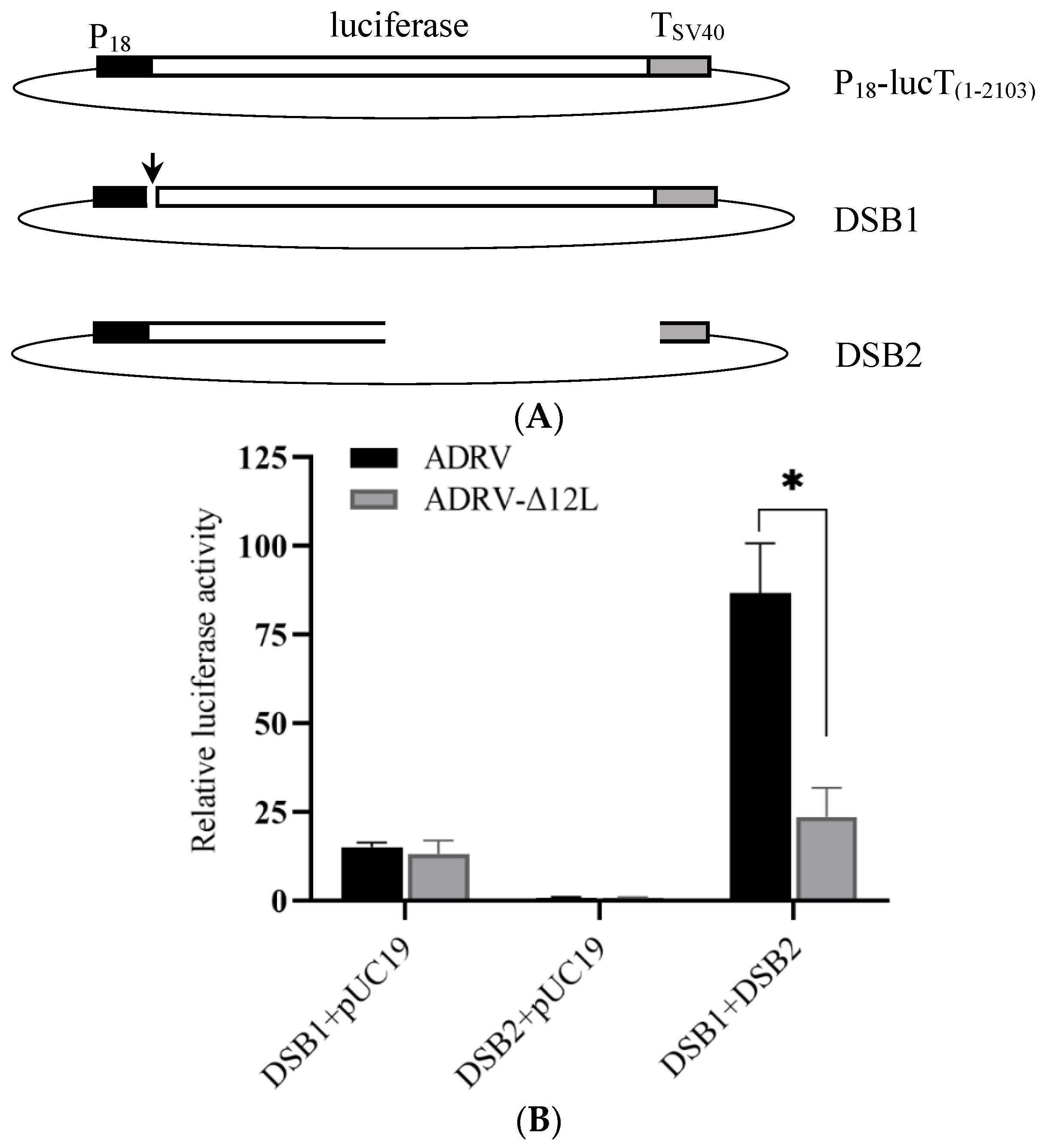
3.4. Deletion of 12L Impaired DNA Homologous Recombination and Double-Stranded Break Repair
3.5. Deletion of 12L Impaired ADRV Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chinchar, V.G.; Hick, P.; Ince, I.A.; Jancovich, J.K.; Marschang, R.; Qin, Q.; Subramaniam, K.; Waltzek, T.B.; Whittington, R.; Williams, T.; et al. ICTV virus taxonomy profile: Iridoviridae. J. Gen. Virol. 2017, 98, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Price, S.J.; Ariel, E.; Maclaine, A.; Rosa, G.M.; Gray, M.J.; Brunner, J.L.; Garner, T.W.J. From fish to frogs and beyond: Impact and host range of emergent ranaviruses. Virology 2017, 511, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Herath, J.; Ellepola, G.; Meegaskumbura, M. Patterns of infection, origins, and transmission of ranaviruses among the ectothermic vertebrates of Asia. Ecol. Evol. 2021, 11, 15498–15519. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.R.K.; Subramaniam, K.; Chinchar, V.G.; Waltzek, T.B. Genomic Sequencing of Ranavirus Isolates from a Three-Spined Stickleback (Gasterosteus aculeatus) and a Red-Legged Frog (Rana aurora). Microbiol. Resour Announc. 2021, 10, e0090221. [Google Scholar] [CrossRef] [PubMed]
- Chinchar, V.G.; Waltzek, T.B. Ranaviruses: Not just for frogs. PLoS Path. 2014, 10, e1003850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.Y.; Gui, J.F. Virus genomes and virus-host interactions in aquaculture animals. Sci. China Life Sci. 2015, 58, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Gui, L.; Chinchar, V.G.; Zhang, Q.Y. Molecular basis of pathogenesis of emerging viruses infecting aquatic animals. Aquac. Fish. 2018, 3, 1–5. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Gui, J.F.; Gao, X.C.; Pei, C.; Hong, Y.J.; Zhang, Q.Y. Genome architecture changes and major gene variations of Andrias davidianus ranavirus (ADRV). Vet. Res. 2013, 44, 101. [Google Scholar] [CrossRef] [Green Version]
- Grasby, J.A.; Finger, L.D.; Tsutakawa, S.E.; Atack, J.M.; Tainer, J.A. Unpairing and gating: Sequence-independent substrate recognition by FEN superfamily nucleases. Trends Biochem. Sci. 2012, 37, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Tomkinson, A.E.; Bardwell, A.J.; Bardwell, L.; Tappe, N.J.; Friedberg, E.C. Yeast DNA-Repair and Recombination Proteins Rad1 and Rad10 Constitute a Single-Stranded-DNA Endonuclease. Nature 1993, 362, 860–862. [Google Scholar] [CrossRef]
- Jimeno, S.; Herrera-Moyano, E.; Ortega, P.; Aguilera, A. Differential effect of the overexpression of Rad2/XPG family endonucleases on genome integrity in yeast and human cells. DNA Repair 2017, 57, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Gotham, V.J.B.; Ciani, B.; Grasby, J.A. A conserved loop-wedge motif moderates reaction site search and recognition by FEN1. Nucleic Acids Res. 2018, 46, 7858–7872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mietus, M.; Nowak, E.; Jaciuk, M.; Kustosz, P.; Studnicka, J.; Nowotny, M. Crystal structure of the catalytic core of Rad2: Insights into the mechanism of substrate binding. Nucleic Acids Res. 2014, 42, 10762–10775. [Google Scholar] [CrossRef] [Green Version]
- Klungland, A.; Lindahl, T. Second pathway for completion of human DNA base excision-repair: Reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 1997, 16, 3341–3348. [Google Scholar] [CrossRef] [Green Version]
- Song, B.; Hamdan, S.M.; Hingorani, M.M. Positioning the 5’-flap junction in the active site controls the rate of flap endonuclease-1-catalyzed DNA cleavage. J. Biol. Chem. 2018, 293, 4792–4804. [Google Scholar] [CrossRef] [Green Version]
- Senkevich, T.G.; Koonin, E.V.; Moss, B. Predicted poxvirus FEN1-like nuclease required for homologous recombination, double-strand break repair and full-size genome formation. Proc. Natl. Acad. Sci. USA 2009, 106, 17921–17926. [Google Scholar] [CrossRef] [Green Version]
- Majji, S.; Thodima, V.; Sample, R.; Whitley, D.; Deng, Y.; Mao, J.; Chinchar, V.G. Transcriptome analysis of Frog virus 3, the type species of the genus Ranavirus, family Iridoviridae. Virology 2009, 391, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.Y.; Ou, T.; Zhu, R.L.; Zhang, Q.Y. Sequencing and analysis of the complete genome of Rana grylio virus (RGV). Arch. Virol. 2012, 157, 1559–1564. [Google Scholar] [CrossRef]
- Mavian, C.; Lopez-Bueno, A.; Balseiro, A.; Casais, R.; Alcami, A.; Alejo, A. The genome sequence of the emerging common midwife toad virus identifies an evolutionary intermediate within ranaviruses. J. Virol. 2012, 86, 3617–3625. [Google Scholar] [CrossRef] [Green Version]
- Ke, F.; Yu, X.-D.; Wang, Z.-H.; Gui, J.-F.; Zhang, Q.-Y. Replication and transcription machinery for ranaviruses: Components, correlation, and functional architecture. Cell Biosci. 2022, 12, 6. [Google Scholar] [CrossRef]
- Yuan, J.D.; Chen, Z.Y.; Huang, X.; Gao, X.C.; Zhang, Q.Y. Establishment of three cell lines from Chinese giant salamander and their sensitivities to the wild-type and recombinant ranavirus. Vet. Res. 2015, 46, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, F.; Wang, Z.H.; Ming, C.Y.; Zhang, Q.Y. Ranaviruses bind cells from different species through interaction with heparan sulfate. Viruses 2019, 11, 593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- He, L.B.; Ke, F.; Wang, J.; Gao, X.C.; Zhang, Q.Y. Rana grylio virus (RGV) envelope protein 2L: Subcellular localization and essential roles in virus infectivity revealed by conditional lethal mutant. J. Gen. Virol. 2014, 95, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Chen, Z.Y.; Wang, J.; Yuan, J.D.; Liao, X.Y.; Gui, J.F.; Zhang, Q.Y. Extensive diversification of MHC in Chinese giant salamanders Andrias davidianus (Anda-MHC) reveals novel splice variants. Dev. Comp. Immunol. 2014, 42, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ke, F.; Huang, Y.H.; Zhao, J.G.; Gui, J.F.; Zhang, Q.Y. Identification and characterization of a novel envelope protein in Rana grylio virus. J. Gen. Virol. 2008, 89, 1866–1872. [Google Scholar] [CrossRef]
- Zeng, X.T.; Gao, X.C.; Zhang, Q.Y. Rana grylio virus 43R encodes an envelope protein involved in virus entry. Virus Genes 2018, 54, 779–791. [Google Scholar] [CrossRef]
- He, L.B.; Ke, F.; Zhang, Q.Y. Rana grylio virus as a vector for foreign gene expression in fish cells. Virus Res. 2012, 163, 66–73. [Google Scholar] [CrossRef]
- Parks, R.J.; Winchcombe-Forhan, C.; DeLange, A.M.; Xing, X.; Evans, D.H. DNA ligase gene disruptions can depress viral growth and replication in poxvirus-infected cells. Virus Res. 1998, 56, 135–147. [Google Scholar] [CrossRef]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef] [Green Version]
- Wang, I.H.; Suomalainen, M.; Andriasyan, V.; Kilcher, S.; Mercer, J.; Neef, A.; Luedtke, N.W.; Greber, U.F. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe 2013, 14, 468–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammon, D.B.; Evans, D.H. The 3’-to-5’ Exonuclease Activity of Vaccinia Virus DNA Polymerase Is Essential and Plays a Role in Promoting Virus Genetic Recombination. J. Virol. 2009, 83, 4236–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culyba, M.J.; Minkah, N.; Hwang, Y.; Benhamou, O.M.J.; Bushman, F.D. DNA branch nuclease activity of vaccinia A22 resolvase. J. Biol. Chem. 2007, 282, 34644–34652. [Google Scholar] [CrossRef] [Green Version]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Piazza, A.; Heyer, W.D. Homologous Recombination and the Formation of Complex Genomic Rearrangements. Trends Cell Biol. 2019, 29, 135–149. [Google Scholar] [CrossRef]
- He, L.B.; Gao, X.C.; Ke, F.; Zhang, Q.Y. A conditional lethal mutation in Rana grylio virus ORF 53R resulted in a marked reduction in virion formation. Virus Res. 2013, 177, 194–200. [Google Scholar] [CrossRef]
- Andino Fde, J.; Grayfer, L.; Chen, G.; Chinchar, V.G.; Edholm, E.S.; Robert, J. Characterization of Frog Virus 3 knockout mutants lacking putative virulence genes. Virology 2015, 485, 162–170. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Deng, L.; Geng, Y.; Zhao, R.; Gray, M.J.; Wang, K.; Ouyang, P.; Chen, D.; Huang, X.; Chen, Z.; Huang, C.; et al. CMTV-like ranavirus infection associated with high mortality in captive catfish-like loach, Triplophysa siluorides, in China. Transbound Emerg. Dis. 2020, 67, 1330–1335. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, F.; Zhang, Q.-Y. ADRV 12L: A Ranaviral Putative Rad2 Family Protein Involved in DNA Recombination and Repair. Viruses 2022, 14, 908. https://doi.org/10.3390/v14050908
Ke F, Zhang Q-Y. ADRV 12L: A Ranaviral Putative Rad2 Family Protein Involved in DNA Recombination and Repair. Viruses. 2022; 14(5):908. https://doi.org/10.3390/v14050908
Chicago/Turabian StyleKe, Fei, and Qi-Ya Zhang. 2022. "ADRV 12L: A Ranaviral Putative Rad2 Family Protein Involved in DNA Recombination and Repair" Viruses 14, no. 5: 908. https://doi.org/10.3390/v14050908
APA StyleKe, F., & Zhang, Q. -Y. (2022). ADRV 12L: A Ranaviral Putative Rad2 Family Protein Involved in DNA Recombination and Repair. Viruses, 14(5), 908. https://doi.org/10.3390/v14050908