
Cardiovascular Tropism and Sequelae of SARS-CoV-2 Infection


{kind=link}
{kind=link}
Abstract
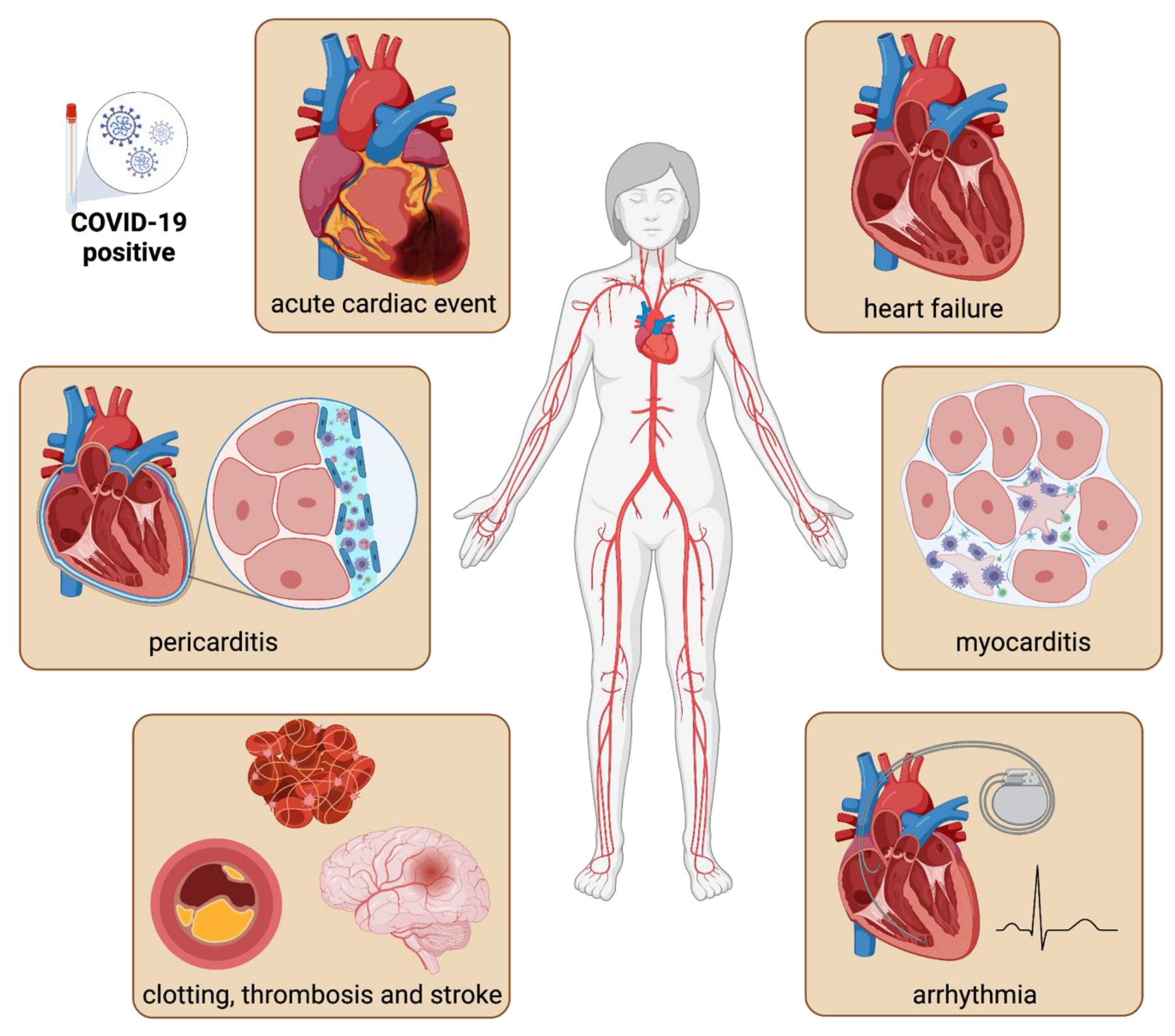
:1. Epidemiology of Cardiovascular COVID-19 Manifestations
2. Pathophysiology
3. SARS-CoV-2 Tropism within the Cardiovascular System
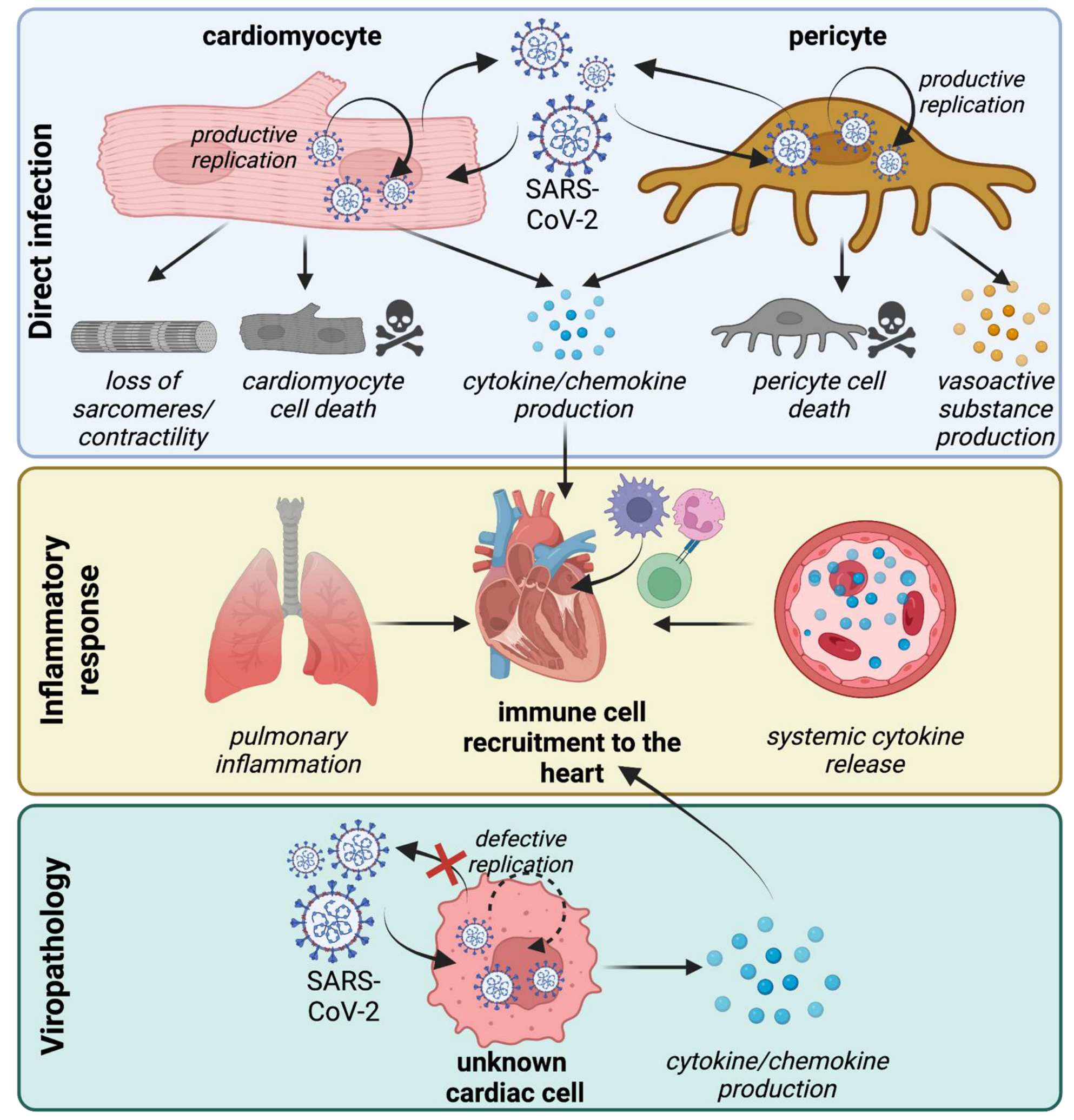
4. Mechanisms of Cardiomyocyte Infection
5. Mechanisms of Pericyte Infection
6. Non-Infectious Effects of SARS-CoV-2 Virions
7. Outstanding Questions and Future Directions
- How does SARS-CoV-2 spread to the heart? SARS-CoV-2 infection originates in the lung and disseminates to multiple organs in the body, including the heart. It is not clear if cardiac SARS-CoV-2 infection occurs through transient viremia, immune cell trafficking, or by exposure to the virus originating from the pleural and/or pericardial spaces.
- Do cardiomyocytes and pericytes express co-receptors for viral entry in addition to ACE2? Multiple alternate receptors for SARS-CoV-2 have been identified in cell lines of various origins, including CD147 [122], LFA-1 [123], and heparan sulfate [124]. The role of these receptors in cardiac infection remains unknown.
- What is the role of cardiomyocyte syncytia formation in clinical disease? Both viral infection and restricted spike protein expression have been shown to cause syncytia formation between hPSC-derived cardiomyocytes. However, syncytia have rarely been observed in human heart specimens and their contribution to cardiac dysfunction remains to be investigated.
- Are there intrinsic defense mechanisms that protect host cardiomyocytes from infection? While SARS-CoV-2 causes robust infection and cell death of cardiomyocytes in vitro, cardiac infection and injury in human specimens appear more restricted. These observations suggest the existence of restriction factors that may limit viral entry and replication or promote viral clearance and cell survival.
- What is the role of direct cardiomyocyte and pericyte infection in acute and post-acute COVID-19 disease progression? The relative contribution of cardiomyocyte and pericyte infection to cardiac pathology is unclear. Animal models with restricted viral receptor expression may help separate their respective contributions.
- Does viropathology contribute to cardiovascular complications of COVID-19? Macrophages and endothelial cells do not support SARS-CoV-2 replication. However, they may become activated by structural components of SARS-CoV-2 and contribute to organ dysfunction. Immune activation and inflammation without direct viral infection contribute to cardiovascular phenotypes. Animal models will be crucial to dissect this question.
- Do variants of concern contribute differently to the progression of cardiac COVID-19? SARS-CoV-2 variants of concern (VOC) harbor mutations that allow them to be transmitted more easily and potentially increase disease severity. While VOC have been shown to infect cardiac cells, it is not clear if they lead to worsened cardiac pathology or outcomes.
- Does vaccination provide protection against long-term cardiac complications after breakthrough infections? Vaccines have changed the course of the pandemic by dramatically decreasing the transmission and disease severity of COVID-19. However, breakthrough infections still occur in vaccinated individuals. It is unclear if the risks and mechanisms of cardiac complications that follow these breakthrough infections are different from those in unvaccinated cases.
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- The Johns Hopkins University School of Medicine. COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://publichealthupdate.com/jhu/ (accessed on 2 April 2022).
- Dubey, A.; Choudhary, S.; Kumar, P.; Tomar, S. Emerging SARS-CoV-2 Variants: Genetic Variability and Clinical Implications. Curr. Microbiol. 2021, 79, 20. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Burkert, F.R.; Lanser, L.; Bellmann-Weiler, R.; Weiss, G. Coronavirus Disease 2019: Clinics, Treatment, and Prevention. Front. Microbiol. 2021, 12, 761887. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.T.; Leung, K.; Bushman, M.; Kishore, N.; Niehus, R.; de Salazar, P.M.; Cowling, B.J.; Lipsitch, M.; Leung, G.M. Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China. Nat. Med. 2020, 26, 506–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 1031. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Qu, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Office for National Statistics. Coronavirus (COVID-19) Related Deaths by Ethnic Group, England and Wales: 2 March 2020 to 10 April 2020. 2020. Available online: https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/articles/coronavirusrelateddeathsbyethnicgroupenglandandwales/2march2020to10april2020 (accessed on 2 April 2022).
- Zheng, Y.Y.; Ma, Y.T.; Zhang, J.Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [Green Version]
- Arentz, M.; Yim, E.; Klaff, L.; Lokhandwala, S.; Riedo, F.X.; Chong, M.; Lee, M. Characteristics and Outcomes of 21 Critically Ill Patients With COVID-19 in Washington State. JAMA 2020, 323, 1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, P.; Choi, J.J.; Pinheiro, L.C.; Schenck, E.J.; Chen, R.; Jabri, A.; Satlin, M.J.; Campion, T.R., Jr.; Nahid, M.; Ringel, J.B.; et al. Clinical Characteristics of Covid-19 in New York City. N. Engl. J. Med. 2020, 382, 2372–2374. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Hou, K.; Xu, R.; Li, Z.; Fu, H.; Wen, L.; Xie, L.; Liu, H.; Selvanayagam, J.B.; Zhang, N.; et al. Clinical Characteristics and Risk Factors of Cardiac Involvement in COVID-19. J. Am. Heart Assoc. 2020, 9, e016807. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; Bonelli, A.; Caronna, A.; Conforti, G.; Iovine, M.; Carbone, A.; Berretta, M.; Botti, G.; Maurea, N. SARS-CoV-2 Infection and Cardioncology: From Cardiometabolic Risk Factors to Outcomes in Cancer Patients. Cancers 2020, 12, 3316. [Google Scholar] [CrossRef]
- Zeng, J.H.; Liu, Y.X.; Yuan, J.; Wang, F.X.; Wu, W.B.; Li, J.X.; Wang, L.F.; Gao, H.; Wang, Y.; Dong, C.F.; et al. First case of COVID-19 complicated with fulminant myocarditis: A case report and insights. Infection 2020, 48, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Lavie, C.J.; Sanchis-Gomar, F. Cardiac troponin I in patients with coronavirus disease 2019 (COVID-19): Evidence from a meta-analysis. Prog. Cardiovasc. Dis. 2020, 63, 390–391. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802. [Google Scholar] [CrossRef] [Green Version]
- Inciardi, R.M.; Lupi, L.; Zaccone, G.; Italia, L.; Raffo, M.; Tomasoni, D.; Cani, D.S.; Cerini, M.; Farina, D.; Gavazzi, E.; et al. Cardiac Involvement in a Patient With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 819–824. [Google Scholar] [CrossRef] [Green Version]
- Stefanini, G.G.; Montorfano, M.; Trabattoni, D.; Andreini, D.; Ferrante, G.; Ancona, M.; Metra, M.; Curello, S.; Maffeo, D.; Pero, G.; et al. ST-Elevation Myocardial Infarction in Patients with COVID-19: Clinical and Angiographic Outcomes. Circulation 2020, 141, 2113–2116. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Bangalore, S.; Sharma, A.; Slotwiner, A.; Yatskar, L.; Harari, R.; Shah, B.; Ibrahim, H.; Friedman, G.H.; Thompson, C.; Alviar, C.L.; et al. ST-Segment Elevation in Patients with Covid-19—A Case Series. N. Engl. J. Med. 2020, 382, 2478–2480. [Google Scholar] [CrossRef] [PubMed]
- Fried, J.A.; Ramasubbu, K.; Bhatt, R.; Topkara, V.K.; Clerkin, K.J.; Horn, E.; Rabbani, L.; Brodie, D.; Jain, S.S.; Kirtane, A.; et al. The Variety of Cardiovascular Presentations of COVID-19. Circulation 2020, 141, 1930–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiezia, L.; Boscolo, A.; Poletto, F.; Cerruti, L.; Tiberio, I.; Campello, E.; Navalesi, P.; Simioni, P. COVID-19-Related Severe Hypercoagulability in Patients Admitted to Intensive Care Unit for Acute Respiratory Failure. Thromb. Haemost. 2020, 120, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [Green Version]
- Puntmann, V.O.; Carerj, M.L.; Wieters, I.; Fahim, M.; Arendt, C.; Hoffmann, J.; Shchendrygina, A.; Escher, F.; Vasa-Nicotera, M.; Zeiher, A.M.; et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 1265–1273. [Google Scholar] [CrossRef]
- Huang, L.; Zhao, P.; Tang, D.; Zhu, T.; Han, R.; Zhan, C.; Liu, W.; Zeng, H.; Tao, Q.; Xia, L. Cardiac Involvement in Patients Recovered From COVID-2019 Identified Using Magnetic Resonance Imaging. JACC Cardiovasc. Imaging 2020, 13, 2330–2339. [Google Scholar] [CrossRef]
- Katsoularis, I.; Fonseca-Rodriguez, O.; Farrington, P.; Lindmark, K.; Fors Connolly, A.M. Risk of acute myocardial infarction and ischaemic stroke following COVID-19 in Sweden: A self-controlled case series and matched cohort study. Lancet 2021, 398, 599–607. [Google Scholar] [CrossRef]
- Bilaloglu, S.; Aphinyanaphongs, Y.; Jones, S.; Iturrate, E.; Hochman, J.; Berger, J.S. Thrombosis in Hospitalized Patients With COVID-19 in a New York City Health System. JAMA 2020, 324, 799–801. [Google Scholar] [CrossRef]
- Kunutsor, S.K.; Laukkanen, J.A. Incidence of venous and arterial thromboembolic complications in COVID-19: A systematic review and meta-analysis. Thromb. Res. 2020, 196, 27–30. [Google Scholar] [CrossRef]
- Middeldorp, S.; Coppens, M.; van Haaps, T.F.; Foppen, M.; Vlaar, A.P.; Muller, M.C.A.; Bouman, C.C.S.; Beenen, L.F.M.; Kootte, R.S.; Heijmans, J.; et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1995–2002. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.; Campia, U.; Hurwitz, S.; Snyder, J.E.; Rizzo, S.M.; Pfeferman, M.B.; Morrison, R.B.; Leiva, O.; Fanikos, J.; Nauffal, V.; et al. Registry of Arterial and Venous Thromboembolic Complications in Patients With COVID-19. J. Am. Coll. Cardiol. 2020, 76, 2060–2072. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H.; Levi, M.; Thachil, J. Coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Poletto, F.; Spiezia, L.; Simion, C.; Campello, E.; Dalla Valle, F.; Tormene, D.; Camporese, G.; Simioni, P. Risk Factors of Venous Thromboembolism in Noncritically Ill Patients Hospitalized for Acute COVID-19 Pneumonia Receiving Prophylactic-Dose Anticoagulation. Viruses 2022, 14, 737. [Google Scholar] [CrossRef]
- Rahi, M.S.; Jindal, V.; Reyes, S.P.; Gunasekaran, K.; Gupta, R.; Jaiyesimi, I. Hematologic disorders associated with COVID-19: A review. Ann. Hematol. 2021, 100, 309–320. [Google Scholar] [CrossRef]
- Carfi, A.; Bernabei, R.; Landi, F. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef]
- Johansson, M.; Stahlberg, M.; Runold, M.; Nygren-Bonnier, M.; Nilsson, J.; Olshansky, B.; Bruchfeld, J.; Fedorowski, A. Long-Haul Post-COVID-19 Symptoms Presenting as a Variant of Postural Orthostatic Tachycardia Syndrome: The Swedish Experience. JACC Case Rep. 2021, 3, 573–580. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Yong, S.J. Long COVID or post-COVID-19 syndrome: Putative pathophysiology, risk factors, and treatments. Infect. Dis. 2021, 53, 737–754. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, E.; Bowe, B.; Al-Aly, Z. Long-term cardiovascular outcomes of COVID-19. Nat. Med. 2022, 28, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, M.; Nazari, M. New Generation Vaccines for COVID-19 Based on Peptide, Viral Vector, Artificial Antigen Presenting Cell, DNA or mRNA. Avicenna J. Med. Biotechnol. 2022, 14, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Plumb, I.D.; Feldstein, L.R.; Barkley, E.; Posner, A.B.; Bregman, H.S.; Hagen, M.B.; Gerhart, J.L. Effectiveness of COVID-19 mRNA Vaccination in Preventing COVID-19-Associated Hospitalization Among Adults with Previous SARS-CoV-2 Infection—United States, June 2021-February 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.M.; Vostok, J.; Johnson, H.; Burns, M.; Gharpure, R.; Sami, S.; Sabo, R.T.; Hall, N.; Foreman, A.; Schubert, P.L.; et al. Outbreak of SARS-CoV-2 Infections, Including COVID-19 Vaccine Breakthrough Infections, Associated with Large Public Gatherings—Barnstable County, Massachusetts, July 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 1059–1062. [Google Scholar] [CrossRef]
- Eggink, D.; Andeweg, S.P.; Vennema, H.; van Maarseveen, N.; Vermaas, K.; Vlaemynck, B.; Schepers, R.; van Gageldonk-Lafeber, A.B.; van den Hof, S.; Reusken, C.B.; et al. Increased risk of infection with SARS-CoV-2 Omicron BA.1 compared with Delta in vaccinated and previously infected individuals, the Netherlands, 22 November 2021 to 19 January 2022. Eurosurveill 2022, 27, 2101196. [Google Scholar] [CrossRef]
- Witberg, G.; Barda, N.; Hoss, S.; Richter, I.; Wiessman, M.; Aviv, Y.; Grinberg, T.; Auster, O.; Dagan, N.; Balicer, R.D.; et al. Myocarditis after Covid-19 Vaccination in a Large Health Care Organization. N. Engl. J. Med. 2021, 385, 2132–2139. [Google Scholar] [CrossRef]
- Husby, A.; Hansen, J.V.; Fosbol, E.; Thiesson, E.M.; Madsen, M.; Thomsen, R.W.; Sorensen, H.T.; Andersen, M.; Wohlfahrt, J.; Gislason, G.; et al. SARS-CoV-2 vaccination and myocarditis or myopericarditis: Population based cohort study. BMJ 2021, 375, e068665. [Google Scholar] [CrossRef]
- Bozkurt, B.; Kamat, I.; Hotez, P.J. Myocarditis With COVID-19 mRNA Vaccines. Circulation 2021, 144, 471–484. [Google Scholar] [CrossRef]
- Verma, A.K.; Lavine, K.J.; Lin, C.Y. Myocarditis after Covid-19 mRNA Vaccination. N. Engl. J. Med. 2021, 385, 1332–1334. [Google Scholar] [CrossRef]
- Block, J.P.; Boehmer, T.K.; Forrest, C.B.; Carton, T.W.; Lee, G.M.; Ajani, U.A.; Christakis, D.A.; Cowell, L.G.; Draper, C.; Ghildayal, N.; et al. Cardiac Complications After SARS-CoV-2 Infection and mRNA COVID-19 Vaccination—PCORnet, United States, January 2021-January 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 517–523. [Google Scholar] [CrossRef]
- Galani, I.E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Mudd, P.A.; Crawford, J.C.; Turner, J.S.; Souquette, A.; Reynolds, D.; Bender, D.; Bosanquet, J.P.; Anand, N.J.; Striker, D.A.; Martin, R.S.; et al. Distinct inflammatory profiles distinguish COVID-19 from influenza with limited contributions from cytokine storm. Sci. Adv. 2020, 6, eabe3024. [Google Scholar] [CrossRef] [PubMed]
- Holter, J.C.; Pischke, S.E.; de Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.M.; Magomedov, A.; Kurreck, A.; Munch, F.H.; Koerner, R.; Kamhieh-Milz, J.; Kahl, A.; Gotthardt, I.; Piper, S.K.; Eckardt, K.U.; et al. Thromboembolic complications in critically ill COVID-19 patients are associated with impaired fibrinolysis. Crit. Care 2020, 24, 676. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef]
- Melillo, F.; Napolano, A.; Loffi, M.; Regazzoni, V.; Boccellino, A.; Danzi, G.B.; Cappelletti, A.M.; Rovere-Querini, P.; Landoni, G.; Ingallina, G.; et al. Myocardial injury in patients with SARS-CoV-2 pneumonia: Pivotal role of inflammation in COVID-19. Eur. J. Clin. Investig. 2022, 52, e13703. [Google Scholar] [CrossRef]
- Clark, D.E.; Dendy, J.M.; Li, D.L.; Crum, K.; Dixon, D.; George-Durrett, K.; Parikh, A.P.; Wassenaar, J.W.; Hughes, S.G.; Soslow, J.H. Cardiovascular magnetic resonance evaluation of soldiers after recovery from symptomatic SARS-CoV-2 infection: A case-control study of cardiovascular post-acute sequelae of SARS-CoV-2 infection (CV PASC). J. Cardiovasc. Magn. Reson. 2021, 23, 106. [Google Scholar] [CrossRef]
- Nagele, M.P.; Haubner, B.; Tanner, F.C.; Ruschitzka, F.; Flammer, A.J. Endothelial dysfunction in COVID-19: Current findings and therapeutic implications. Atherosclerosis 2020, 314, 58–62. [Google Scholar] [CrossRef]
- Bisaccia, G.; Ricci, F.; Recce, V.; Serio, A.; Iannetti, G.; Chahal, A.A.; Stahlberg, M.; Khanji, M.Y.; Fedorowski, A.; Gallina, S. Post-Acute Sequelae of COVID-19 and Cardiovascular Autonomic Dysfunction: What Do We Know? J. Cardiovasc. Dev. Dis. 2021, 8, 156. [Google Scholar] [CrossRef]
- Stahlberg, M.; Reistam, U.; Fedorowski, A.; Villacorta, H.; Horiuchi, Y.; Bax, J.; Pitt, B.; Matskeplishvili, S.; Luscher, T.F.; Weichert, I.; et al. Post-COVID-19 Tachycardia Syndrome: A Distinct Phenotype of Post-Acute COVID-19 Syndrome. Am. J. Med. 2021, 134, 1451–1456. [Google Scholar] [CrossRef]
- Basso, C.; Leone, O.; Rizzo, S.; De Gaspari, M.; van der Wal, A.C.; Aubry, M.C.; Bois, M.C.; Lin, P.T.; Maleszewski, J.J.; Stone, J.R. Pathological features of COVID-19-associated myocardial injury: A multicentre cardiovascular pathology study. Eur. Heart J. 2020, 41, 3827–3835. [Google Scholar] [CrossRef] [PubMed]
- Bearse, M.; Hung, Y.P.; Krauson, A.J.; Bonanno, L.; Boyraz, B.; Harris, C.K.; Helland, T.L.; Hilburn, C.F.; Hutchison, B.; Jobbagy, S.; et al. Factors associated with myocardial SARS-CoV-2 infection, myocarditis, and cardiac inflammation in patients with COVID-19. Mod. Pathol. 2021, 34, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Bois, M.C.; Boire, N.A.; Layman, A.J.; Aubry, M.C.; Alexander, M.P.; Roden, A.C.; Hagen, C.E.; Quinton, R.A.; Larsen, C.; Erben, Y.; et al. COVID-19-Associated Nonocclusive Fibrin Microthrombi in the Heart. Circulation 2021, 143, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, D.; Kawakami, R.; Guagliumi, G.; Sakamoto, A.; Kawai, K.; Gianatti, A.; Nasr, A.; Kutys, R.; Guo, L.; Cornelissen, A.; et al. Microthrombi as a Major Cause of Cardiac Injury in COVID-19: A Pathologic Study. Circulation 2021, 143, 1031–1042. [Google Scholar] [CrossRef]
- Lindner, D.; Fitzek, A.; Brauninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.P.; et al. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Bailey, A.L.; Dmytrenko, O.; Greenberg, L.; Bredemeyer, A.L.; Ma, P.; Liu, J.; Penna, V.; Winkler, E.S.; Sviben, S.; Brooks, E.; et al. SARS-CoV-2 Infects Human Engineered Heart Tissues and Models COVID-19 Myocarditis. JACC Basic Transl. Sci. 2021, 6, 331–345. [Google Scholar] [CrossRef]
- Perez-Bermejo, J.A.; Kang, S.; Rockwood, S.J.; Simoneau, C.R.; Joy, D.A.; Silva, A.C.; Ramadoss, G.N.; Flanigan, W.R.; Fozouni, P.; Li, H.; et al. SARS-CoV-2 infection of human iPSC-derived cardiac cells reflects cytopathic features in hearts of patients with COVID-19. Sci. Transl. Med. 2021, 13, eabf7872. [Google Scholar] [CrossRef]
- Tavazzi, G.; Pellegrini, C.; Maurelli, M.; Belliato, M.; Sciutti, F.; Bottazzi, A.; Sepe, P.A.; Resasco, T.; Camporotondo, R.; Bruno, R.; et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur. J. Heart Fail. 2020, 22, 911–915. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; Paul van Schayck, J.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e907. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffre, F.; et al. A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 2020, 27, 125–136.e127. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, N.R.; Chaffin, M.; Bedi, K.C., Jr.; Papangeli, I.; Akkad, A.D.; Arduini, A.; Hayat, S.; Eraslan, G.; Muus, C.; Bhattacharyya, R.P.; et al. Myocyte-Specific Upregulation of ACE2 in Cardiovascular Disease: Implications for SARS-CoV-2-Mediated Myocarditis. Circulation 2020, 142, 708–710. [Google Scholar] [CrossRef] [PubMed]
- McCracken, I.R.; Saginc, G.; He, L.; Huseynov, A.; Daniels, A.; Fletcher, S.; Peghaire, C.; Kalna, V.; Andaloussi-Mae, M.; Muhl, L.; et al. Lack of Evidence of Angiotensin-Converting Enzyme 2 Expression and Replicative Infection by SARS-CoV-2 in Human Endothelial Cells. Circulation 2021, 143, 865–868. [Google Scholar] [CrossRef]
- Schimmel, L.; Chew, K.Y.; Stocks, C.J.; Yordanov, T.E.; Essebier, P.; Kulasinghe, A.; Monkman, J.; Dos Santos Miggiolaro, A.F.R.; Cooper, C.; de Noronha, L.; et al. Endothelial cells are not productively infected by SARS-CoV-2. Clin. Transl. Immunol. 2021, 10, e1350. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Choi, S.W.; Shin, J.S.; Park, S.J.; Jung, E.; Park, Y.G.; Lee, J.; Kim, S.J.; Park, H.J.; Lee, J.H.; Park, S.M.; et al. Antiviral activity and safety of remdesivir against SARS-CoV-2 infection in human pluripotent stem cell-derived cardiomyocytes. Antivir. Res. 2020, 184, 104955. [Google Scholar] [CrossRef]
- Li, Y.; Renner, D.M.; Comar, C.E.; Whelan, J.N.; Reyes, H.M.; Cardenas-Diaz, F.L.; Truitt, R.; Tan, L.H.; Dong, B.; Alysandratos, K.D.; et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2022643118. [Google Scholar] [CrossRef]
- Marchiano, S.; Hsiang, T.Y.; Khanna, A.; Higashi, T.; Whitmore, L.S.; Bargehr, J.; Davaapil, H.; Chang, J.; Smith, E.; Ong, L.P.; et al. SARS-CoV-2 Infects Human Pluripotent Stem Cell-Derived Cardiomyocytes, Impairing Electrical and Mechanical Function. Stem Cell Rep. 2021, 16, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Navaratnarajah, C.K.; Pease, D.R.; Halfmann, P.J.; Taye, B.; Barkhymer, A.; Howell, K.G.; Charlesworth, J.E.; Christensen, T.A.; Kawaoka, Y.; Cattaneo, R.; et al. Highly Efficient SARS-CoV-2 Infection of Human Cardiomyocytes: Spike Protein-Mediated Cell Fusion and Its Inhibition. J. Virol. 2021, 95, e0136821. [Google Scholar] [CrossRef]
- Yang, L.; Nilsson-Payant, B.E.; Han, Y.; Jaffre, F.; Zhu, J.; Wang, P.; Zhang, T.; Redmond, D.; Houghton, S.; Moller, R.; et al. Cardiomyocytes recruit monocytes upon SARS-CoV-2 infection by secreting CCL2. Stem Cell Rep. 2021, 16, 2274–2288. [Google Scholar] [CrossRef]
- Bojkova, D.; Wagner, J.U.G.; Shumliakivska, M.; Aslan, G.S.; Saleem, U.; Hansen, A.; Luxan, G.; Gunther, S.; Pham, M.D.; Krishnan, J.; et al. SARS-CoV-2 infects and induces cytotoxic effects in human cardiomyocytes. Cardiovasc. Res. 2020, 116, 2207–2215. [Google Scholar] [CrossRef]
- Simmons, G.; Zmora, P.; Gierer, S.; Heurich, A.; Pohlmann, S. Proteolytic activation of the SARS-coronavirus spike protein: Cutting enzymes at the cutting edge of antiviral research. Antivir. Res. 2013, 100, 605–614. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pohlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio 2018, 9, e00221-18. [Google Scholar] [CrossRef] [Green Version]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Gotte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quagliariello, V.; Bonelli, A.; Caronna, A.; Lombari, M.C.; Conforti, G.; Libutti, M.; Iaffaioli, R.V.; Berretta, M.; Botti, G.; Maurea, N. SARS-CoV-2 infection: NLRP3 inflammasome as plausible target to prevent cardiopulmonary complications? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9169–9171. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- Sefik, E.; Qu, R.; Junqueira, C.; Kaffe, E.; Mirza, H.; Zhao, J.; Brewer, J.R.; Han, A.; Steach, H.R.; Israelow, B.; et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 2022. [Google Scholar] [CrossRef]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; de Sa, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Goncalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L.; et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021, 7, 43. [Google Scholar] [CrossRef]
- Guggenheim, A.G.; Wright, K.M.; Zwickey, H.L. Immune Modulation From Five Major Mushrooms: Application to Integrative Oncology. Integr. Med. 2014, 13, 32–44. [Google Scholar]
- Wang, L.; Sievert, D.; Clark, A.E.; Lee, S.; Federman, H.; Gastfriend, B.D.; Shusta, E.V.; Palecek, S.P.; Carlin, A.F.; Gleeson, J.G. A human three-dimensional neural-perivascular ‘assembloid’ promotes astrocytic development and enables modeling of SARS-CoV-2 neuropathology. Nat. Med. 2021, 27, 1600–1606. [Google Scholar] [CrossRef]
- Avolio, E.; Carrabba, M.; Milligan, R.; Kavanagh Williamson, M.; Beltrami, A.P.; Gupta, K.; Elvers, K.T.; Gamez, M.; Foster, R.R.; Gillespie, K.; et al. The SARS-CoV-2 Spike protein disrupts human cardiac pericytes function through CD147 receptor-mediated signalling: A potential non-infective mechanism of COVID-19 microvascular disease. Clin. Sci. 2021, 135, 2667–2689. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.; Zidar, D.A.; Bristow, M.R.; Cameron, S.J.; Chan, T.; Harding, C.V., 3rd; Kwon, D.H.; Singh, T.; Tilton, J.C.; Tsai, E.J.; et al. COVID-19 and Cardiovascular Disease: From Bench to Bedside. Circ. Res. 2021, 128, 1214–1236. [Google Scholar] [CrossRef] [PubMed]
- Volz, K.S.; Jacobs, A.H.; Chen, H.I.; Poduri, A.; McKay, A.S.; Riordan, D.P.; Kofler, N.; Kitajewski, J.; Weissman, I.; Red-Horse, K. Pericytes are progenitors for coronary artery smooth muscle. Elife 2015, 4, e10036. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. Beta-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e1514. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Vignuzzi, M.; Lopez, C.B. Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol. 2019, 4, 1075–1087. [Google Scholar] [CrossRef]
- Anand, G.; Perry, A.M.; Cummings, C.L.; St Raymond, E.; Clemens, R.A.; Steed, A.L. Surface Proteins of SARS-CoV-2 Drive Airway Epithelial Cells to Induce IFN-Dependent Inflammation. J. Immunol. 2021, 206, 3000–3009. [Google Scholar] [CrossRef]
- Bi, Z.; Hong, W.; Que, H.; He, C.; Ren, W.; Yang, J.; Lu, T.; Chen, L.; Lu, S.; Peng, X.; et al. Inactivated SARS-CoV-2 induces acute respiratory distress syndrome in human ACE2-transgenic mice. Signal Transduct. Target. Ther. 2021, 6, 439. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.L.; Solopov, P.A.; Sharlow, E.R.; Lazo, J.S.; Marik, P.E.; Catravas, J.D. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Kappa18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L477–L484. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Kizaki, T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021, 31, 818–820. [Google Scholar] [CrossRef] [PubMed]
- Negron, S.G.; Kessinger, C.W.; Xu, B.; Pu, W.T.; Lin, Z. Selectively expressing SARS-CoV-2 Spike protein S1 subunit in cardiomyocytes induces cardiac hypertrophy in mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Francis, M.E.; Goncin, U.; Kroeker, A.; Swan, C.; Ralph, R.; Lu, Y.; Etzioni, A.L.; Falzarano, D.; Gerdts, V.; Machtaler, S.; et al. SARS-CoV-2 infection in the Syrian hamster model causes inflammation as well as type I interferon dysregulation in both respiratory and non-respiratory tissues including the heart and kidney. PLoS Pathog. 2021, 17, e1009705. [Google Scholar] [CrossRef]
- Winkler, E.S.; Chen, R.E.; Alam, F.; Yildiz, S.; Case, J.B.; Uccellini, M.B.; Holtzman, M.J.; Garcia-Sastre, A.; Schotsaert, M.; Diamond, M.S. SARS-CoV-2 causes lung infection without severe disease in human ACE2 knock-in mice. J. Virol. 2021, 96, JVI0151121. [Google Scholar] [CrossRef]
- Sun, S.; Gu, H.; Cao, L.; Chen, Q.; Ye, Q.; Yang, G.; Li, R.T.; Fan, H.; Deng, Y.Q.; Song, X.; et al. Characterization and structural basis of a lethal mouse-adapted SARS-CoV-2. Nat. Commun. 2021, 12, 5654. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, Y.; Hui, X.; Zhao, Y.; Gong, W.; Wang, T.; Zhang, S.; Yang, Y.; Deng, F.; Zhang, Q.; et al. Q493K and Q498H substitutions in Spike promote adaptation of SARS-CoV-2 in mice. EBioMedicine 2021, 67, 103381. [Google Scholar] [CrossRef]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef]
- Shen, X.-R.; Geng, R.; Li, Q.; Chen, Y.; Li, S.-F.; Wang, Q.; Min, J.; Yang, Y.; Li, B.; Jiang, R.-D.; et al. ACE2-independent infection of T lymphocytes by SARS-CoV-2. Signal Transduct. Target. Ther. 2022, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Puray-Chavez, M.; LaPak, K.M.; Schrank, T.P.; Elliott, J.L.; Bhatt, D.P.; Agajanian, M.J.; Jasuja, R.; Lawson, D.Q.; Davis, K.; Rothlauf, P.W.; et al. Systematic analysis of SARS-CoV-2 infection of an ACE2-negative human airway cell. Cell Rep. 2021, 36, 109364. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dmytrenko, O.; Lavine, K.J. Cardiovascular Tropism and Sequelae of SARS-CoV-2 Infection. Viruses 2022, 14, 1137. https://doi.org/10.3390/v14061137
Dmytrenko O, Lavine KJ. Cardiovascular Tropism and Sequelae of SARS-CoV-2 Infection. Viruses. 2022; 14(6):1137. https://doi.org/10.3390/v14061137
Chicago/Turabian StyleDmytrenko, Oleksandr, and Kory J. Lavine. 2022. "Cardiovascular Tropism and Sequelae of SARS-CoV-2 Infection" Viruses 14, no. 6: 1137. https://doi.org/10.3390/v14061137
APA StyleDmytrenko, O., & Lavine, K. J. (2022). Cardiovascular Tropism and Sequelae of SARS-CoV-2 Infection. Viruses, 14(6), 1137. https://doi.org/10.3390/v14061137