
In Vivo and In Vitro Studies of Cigarette Smoke Effects on Innate Responses to Influenza Virus: A Matter of Models?

Abstract
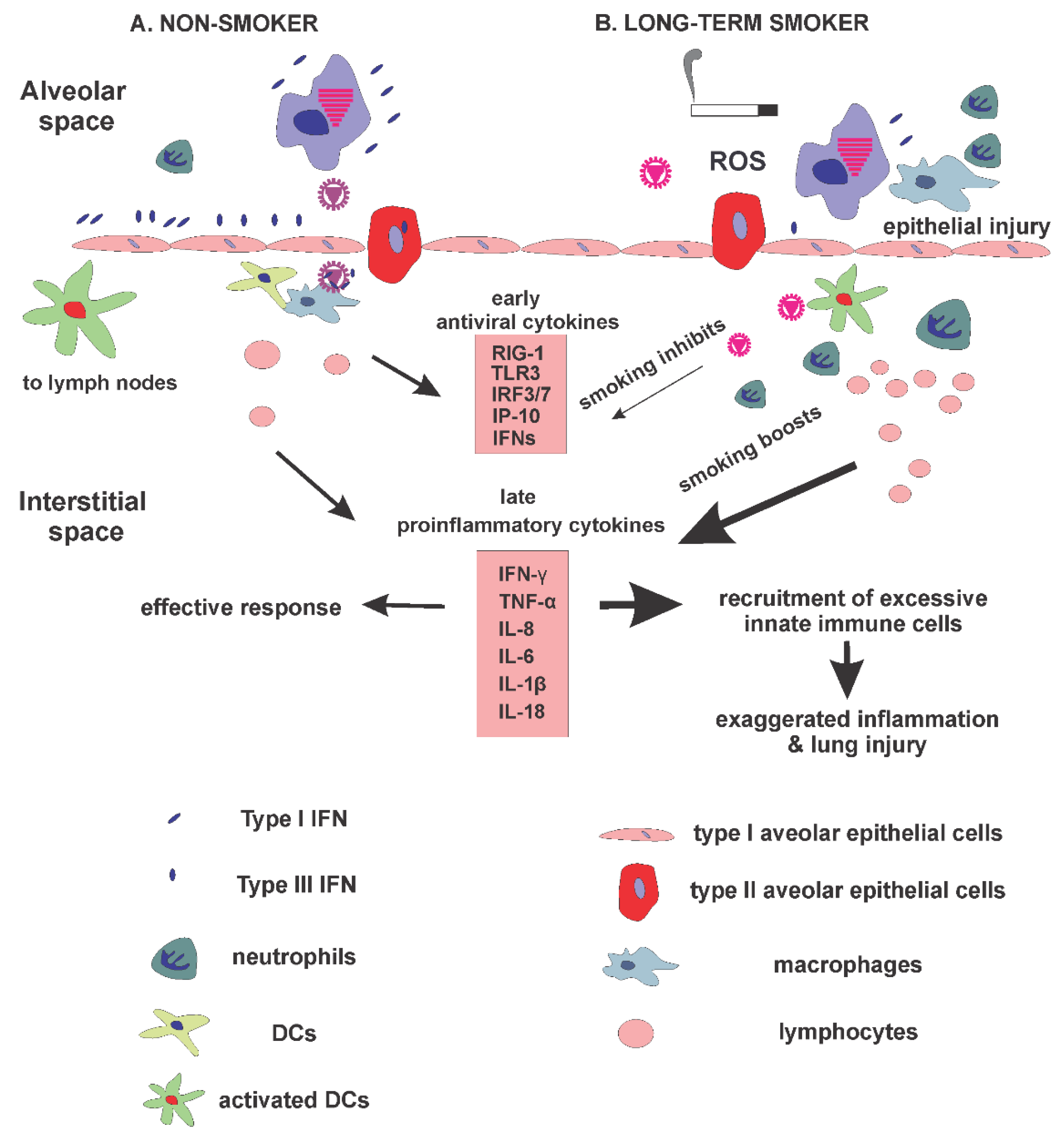
:1. Introduction
2. In Vitro Cell Culture Studies
2.1. Epithelial Cells and Immune Leukocytes in the Innate Immune Response to IAV
2.2. CS Effects on Epithelial Cells during Viral Infection
2.3. CS Effects on Immune Leukocytes during Viral Infection
3. In Vivo Animal Smoking Studies
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Davis, M.M.; Taubert, K.; Benin, A.L.; Brown, D.W.; Mensah, G.A.; Baddour, L.M.; Dunbar, S.; Krumholz, H.M. Influenza vaccination as secondary prevention for cardiovascular disease: A science advisory from the American Heart Association/American College of Cardiology. J. Am. Coll. Cardiol. 2006, 48, 1498–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, R.; Heiden, M.; Offergeld, R.; Burger, R. Influenza Virus. Transfus. Med. Hemotherapy 2009, 36, 32–39. [Google Scholar] [CrossRef]
- Schrauwen, E.J.; de Graaf, M.; Herfst, S.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A.M. Determinants of virulence of influenza A virus. Eur. J. Clin. Microbiol. 2014, 33, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubenberger, J.K.; Morens, D.M. The pathology of influenza virus infections. Annu. Rev. Pathol. 2008, 3, 499–522. [Google Scholar] [CrossRef]
- Nelemans, T.; Kikkert, M. Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections. Viruses 2019, 11, 961. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [Green Version]
- Aoshi, T.; Koyama, S.; Kobiyama, K.; Akira, S.; Ishii, K. Ishii Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 2011, 1, 226–232. [Google Scholar] [CrossRef]
- Koyama, S.; Ishii, K.J.; Coban, C.; Akira, S. Innate immune response to viral infection. Cytokine 2008, 43, 336–341. [Google Scholar] [CrossRef]
- Jung, H.E.; Lee, H.K. Host Protective Immune Responses against Influenza A Virus Infection. Viruses 2020, 12, 504. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, W.; Duggan, E.S.; Booth, J.L.; Zou, M.H.; Metcalf, J.P. RIG-I and TLR3 are both required for maximum interferon induction by influenza virus in human lung alveolar epithelial cells. Virology 2015, 482, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Guillot, L.; Le Goffic, R.; Bloch, S.; Escriou, N.; Akira, S.; Chignard, M.; Si-Tahar, M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 2005, 280, 5571–5580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Metcalf, J.P. The Role of Type I IFNs in Influenza: Antiviral Superheroes or Immunopathogenic Villains? J. Innate Immun. 2020, 12, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomino, D.C.T.; Marti, L.C. Chemokines and immunity. Einstein 2015, 13, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Burney, P.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Prim. 2015, 1, 15076. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Linden, D.; Guo-Parke, H.; Coyle, P.V.; Fairley, D.; McAuley, D.; Taggart, C.C.; Kidney, J. Respiratory viral infection: A potential “missing link” in the pathogenesis of COPD. Eur. Respir. Rev. 2019, 28, 180063. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, M.; Papi, A.; Contoli, M.; Beghé, B.; Celli, B.R.; Wedzicha, J.A.; Fabbri, L.M. Chronic obstructive pulmonary disease exacerbation fundamentals: Diagnosis, treatment, prevention and disease impact. Respirology 2021, 26, 532–551. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, E.R.; Cherniack, R.M. Management of chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2689–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, H.; Hunter, A.; Murray, R.; Lim, W.S.; McKeever, T. Cigarette smoking and the occurrence of influenza-Systematic review. J. Infect. 2019, 79, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Liang, C.-L.; Liu, H.; Zeng, Y.-Q.; Hou, S.; Huang, S.; Lai, X.; Dai, Z. Impacts of cigarette smoking on immune responsiveness: Up and down or upside down? Oncotarget 2017, 8, 268–284. [Google Scholar] [CrossRef] [Green Version]
- Arcavi, L.; Benowitz, N.L. Cigarette smoking and infection. Arch Intern. Med. 2004, 164, 2206–2216. [Google Scholar] [CrossRef]
- Cohen, S.; Tyrrell, A.D.; Russell, A.M.; Jarvis, M.J.; Smith, A.P. Smoking, alcohol consumption, and susceptibility to the common cold. Am. J. Public Health 1993, 83, 1277–1283. [Google Scholar] [CrossRef] [Green Version]
- Blake, G.H.; Abell, T.D.; Stanley, W.G. Cigarette smoking and upper respiratory infection among recruits in basic combat training. Ann. Intern. Med. 1988, 109, 198–202. [Google Scholar] [CrossRef]
- Ambrose, J.A.; Barua, R.S. The pathophysiology of cigarette smoking and cardiovascular disease: An update. J. Am. Coll. Cardiol. 2004, 43, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Sobus, S.L.; Warren, G.W. The biologic effects of cigarette smoke on cancer cells. Cancer 2014, 120, 3617–3626. [Google Scholar] [CrossRef]
- Wang, J.; Oberley-Deegan, R.; Wang, S.; Nikrad, M.; Funk, C.J.; Hartshorn, K.L.; Mason, R.J. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza A infection. J. Immunol. 2009, 182, 1296–1304. [Google Scholar] [CrossRef] [Green Version]
- Ronni, T.; Matikainen, S.; Sareneva, T.; Melen, K.; Pirhonen, J.; Keskinen, P.; Julkunen, I. Regulation of IFN-alpha/beta, MxA, 2’,5’-oligoadenylate synthetase, and HLA gene expression in influenza A-infected human lung epithelial cells. J. Immunol. 1997, 158, 2363–2374. [Google Scholar] [PubMed]
- Julkunen, I.; Melen, K.; Nyqvist, M.; Pirhonen, J.; Sareneva, T.; Matikainen, S. Inflammatory responses in influenza A virus infection. Vaccine 2000, 19 (Suppl. 1), S32–S37. [Google Scholar] [CrossRef]
- Ronni, T.; Sareneva, T.; Pirhonen, J.; Julkunen, I. Activation of IFN-alpha, IFN-gamma, MxA, and IFN regulatory factor 1 genes in influenza A virus-infected human peripheral blood mononuclear cells. J. Immunol. 1995, 154, 2764–2774. [Google Scholar] [PubMed]
- Ioannidis, I.; Ye, F.; McNally, B.; Willette, M.; Flaño, E. Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J. Virol. 2013, 87, 3261–3270. [Google Scholar] [CrossRef] [Green Version]
- Hoeve, M.A.; Nash, A.A.; Jackson, D.; Randall, R.E.; Dransfield, I. Influenza virus A infection of human monocyte and macrophage subpopulations reveals increased susceptibility associated with cell differentiation. PLoS ONE 2012, 7, e29443. [Google Scholar] [CrossRef] [Green Version]
- Kreijtz, J.; Fouchier, R.; Rimmelzwaan, G. Immune responses to influenza virus infection. Virus Res. 2011, 162, 19–30. [Google Scholar] [CrossRef]
- Schleimer, R.P.; Kato, A.; Kern, R.; Kuperman, D.; Avila, P.C. Epithelium: At the interface of innate and adaptive immune responses. J. Allergy Clin. Immunol. 2007, 120, 1279–1284. [Google Scholar] [CrossRef] [Green Version]
- Nicol, M.Q.; Dutia, B.M. The role of macrophages in influenza A virus infection. Future Virol. 2014, 9, 847–862. [Google Scholar] [CrossRef]
- Gill, M.A.; Long, K.; Kwon, T.; Muniz, L.; Mejias, A.; Connolly, J.; Roy, L.; Banchereau, J.; Ramilo, O. Differential Recruitment of Dendritic Cells and Monocytes to Respiratory Mucosal Sites in Children with Influenza Virus or Respiratory Syncytial Virus Infection. J. Infect. Dis. 2008, 198, 1667–1676. [Google Scholar] [CrossRef]
- Dai, J.; Megjugorac, N.J.; Amrute, S.B.; Fitzgerald-Bocarsly, P. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J. Immunol. 2004, 173, 1535–1548. [Google Scholar] [CrossRef] [Green Version]
- Mifsud, E.J.; Kuba, M.; Barr, I.G. Innate Immune Responses to Influenza Virus Infections in the Upper Respiratory Tract. Viruses 2021, 13, 2090. [Google Scholar] [CrossRef] [PubMed]
- Gellner, C.A.; Reynaga, D.D.; Leslie, F.M. Cigarette Smoke Extract: A Preclinical Model of Tobacco Dependence. Curr. Protoc. Neurosci. 2016, 77, 9.54.1–9.54.10. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Patel, K.B.; Booth, J.L.; Zhang, W.; Metcalf, J.P. Cigarette smoke extract suppresses the RIG-I-initiated innate immune response to influenza virus in the human lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L821–L830. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhang, W.; Booth, J.L.; Hutchings, D.C.; Wang, X.; White, V.L.; Youness, H.; Cross, C.D.; Zou, M.H.; Burian, D.; et al. Human primary airway epithelial cells isolated from active smokers have epigenetically impaired antiviral responses. Respir. Res. 2016, 17, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, K.M.; Brighton, L.E.; Zhang, W.; Carson, J.L.; Jaspers, I. Epithelial cells from smokers modify dendritic cell responses in the context of influenza infection. Am. J. Respir. Cell. Mol. Biol. 2011, 45, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaspers, I.; Horvath, K.M.; Zhang, W.; Brighton, L.E.; Carson, J.L.; Noah, T.L. Reduced expression of IRF7 in nasal epithelial cells from smokers after infection with influenza. Am. J. Respir. Cell. Mol. Biol. 2010, 43, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, C.M.; DeWitte-Orr, S.J.; Hornby, K.R.; Zavitz, C.C.; Lichty, B.D.; Stämpfli, M.R.; Mossman, K.L. Cigarette smoke suppresses type I interferon-mediated antiviral immunity in lung fibroblast and epithelial cells. J. Interf. Cytokine Res. 2008, 28, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Danov, O.; Wolff, M.; Bartel, S.; Böhlen, S.; Obernolte, H.; Wronsk, S.I.; Jonigk, D.; Hammer, B.; Kovacevic, D.; Reuter, S.; et al. Cigarette Smoke Affects Dendritic Cell Populations, Epithelial Barrier Function, and the Immune Response to Viral Infection With H1N1. Front. Med. 2020, 7, 571003. [Google Scholar] [CrossRef]
- Duffney, P.F.; McCarthy, C.E.; Nogales, A.; Thatcher, T.H.; Martinez-Sobrido, L.; Phipps, R.P.; Sime, P.J. Cigarette smoke dampens antiviral signaling in small airway epithelial cells by disrupting TLR3 cleavage. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L505–L513. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.; Lamson, D.M.; George, K.S.; Walsh, T.J. Human rhinoviruses. Clin. Microbiol. Rev. 2013, 26, 135–162. [Google Scholar] [CrossRef] [Green Version]
- Hudy, M.H.; Traves, S.L.; Wiehler, S.; Proud, D. Cigarette smoke modulates rhinovirus-induced airway epithelial cell chemokine production. Eur. Respir. J. 2010, 35, 1256–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Kim, H.; Jang, Y.J. Cigarette smoke extract enhances rhinovirus-induced toll-like receptor 3 expression and interleukin-8 secretion in A549 cells. Am. J. Rhinol. Allergy 2009, 23, e5–e9. [Google Scholar] [CrossRef] [PubMed]
- Logan, J.; Chen, L.; Gangell, C.; Sly, P.D.; Fantino, E.; Liu, K. Brief exposure to cigarette smoke impairs airway epithelial cell innate anti-viral defence. Toxicol. In Vitro 2014, 28, 1430–1435. [Google Scholar] [CrossRef]
- Hudy, M.H.; Traves, S.L.; Proud, D. Transcriptional and epigenetic modulation of human rhinovirus-induced CXCL10 production by cigarette smoke. Am. J. Respir. Cell. Mol. Biol. 2014, 50, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Proud, D.; Hudy, M.H.; Wiehler, S.; Zaheer, R.S.; Amin, M.A.; Pelikan, J.B.; Tacon, C.E.; Tonsaker, T.O.; Walker, B.L.; Kooi, C.; et al. Cigarette smoke modulates expression of human rhinovirus-induced airway epithelial host defense genes. PLoS ONE 2012, 7, e40762. [Google Scholar] [CrossRef]
- Pace, E.; Ferraro, M.; Siena, L.; Melis, M.; Montalbano, A.M.; Johnson, M.; Bonsignore, M.R.; Bonsignore, G.; Gjomarkaj, M. Cigarette smoke increases Toll-like receptor 4 and modifies lipopolysaccharide-mediated responses in airway epithelial cells. Immunology 2008, 124, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Hudy, M.H.; Proud, D. Cigarette smoke enhances human rhinovirus-induced CXCL8 production via HuR-mediated mRNA stabilization in human airway epithelial cells. Respir. Res. 2013, 14, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Batra, S. Regulation of DNA methylation signatures on NF-κB and STAT3 pathway genes and TET activity in cigarette smoke extract-challenged cells/COPD exacerbation model in vitro. Cell Biol. Toxicol. 2020, 36, 459–480. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Xie, J.; Zhao, J.; Xu, Y.; Yang, S.; Ni, W. Involvement of Bcl-2 family in apoptosis and signal pathways induced by cigarette smoke extract in the human airway smooth muscle cells. DNA Cell. Biol. 2009, 28, 13–22. [Google Scholar] [CrossRef]
- Love, M.E.; Proud, D. Respiratory Viral and Bacterial Exacerbations of COPD-The Role of the Airway Epithelium. Cells 2022, 11, 1416. [Google Scholar] [CrossRef]
- Mallia, P.; Message, S.D.; Gielen, V.; Contoli, M.; Gray, K.; Kebadze, T.; Aniscenko, J.; Laza-Stanca, V.; Edwards, M.R.; Slater, L.; et al. Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. Am. J. Respir. Crit. Care Med. 2011, 183, 734–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veerati, P.C.; Troy, N.M.; Reid, A.T.; Li, N.F.; Nichol, K.S.; Kaur, P.; Maltby, S.; Wark, P.A.B.; Knight, D.A.; Bosco, A.; et al. Airway Epithelial Cell Immunity Is Delayed During Rhinovirus Infection in Asthma and COPD. Front. Immunol. 2020, 11, 974. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Lazar, Z.; Koenderman, L.; Kraneveld, A.D.; Nijkamp, F.P.; Folkerts, G. Cigarette smoke attenuates the production of cytokines by human plasmacytoid dendritic cells and enhances the release of IL-8 in response to TLR-9 stimulation. Respir. Res. 2009, 10, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroening, P.R.; Barnes, T.W.; Pease, L.; Limper, A.; Kita, H.; Vassallo, R. Cigarette smoke-induced oxidative stress suppresses generation of dendritic cell IL-12 and IL-23 through ERK-dependent pathways. J. Immunol. 2008, 181, 1536–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mian, M.F.; Stämpfli, M.R.; Mossman, K.L.; Ashkar, A.A. Cigarette smoke attenuation of poly I:C-induced innate antiviral responses in human PBMC is mainly due to inhibition of IFN-beta production. Mol. Immunol. 2009, 46, 821–829. [Google Scholar] [CrossRef]
- Metcalfe, H.J.; Lea, S.; Hughes, D.; Khalaf, R.; Abbott-Banner, K.; Singh, D. Effects of cigarette smoke on Toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages. Clin. Exp. Immunol. 2014, 176, 461–472. [Google Scholar] [CrossRef]
- Todt, J.C.; Freeman, C.M.; Brown, J.P.; Sonstein, J.; Ames, T.M.; McCubbrey, A.L.; Martinez, F.J.; Chensue, S.W.; Beck, J.M.; Curtis, J.L. Smoking decreases the response of human lung macrophages to double-stranded RNA by reducing TLR3 expression. Respir. Res. 2013, 14, 33. [Google Scholar] [CrossRef] [Green Version]
- Koarai, A.; Yanagisawa, S.; Sugiura, H.; Ichikawa, T.; Akamatsu, K.; Hirano, T.; Nakanishi, M.; Matsunaga, K.; Minakata, Y.; Ichinose, M. Cigarette smoke augments the expression and responses of toll-like receptor 3 in human macrophages. Respirology 2012, 17, 1018–1025. [Google Scholar] [CrossRef]
- Feng, Y.; Kong, Y.; Barnes, P.F.; Huang, F.-F.; Klucar, P.; Wang, X.; Samten, B.; Sengupta, M.; Machona, B.; Donis, R.; et al. Exposure to cigarette smoke inhibits the pulmonary T-cell response to influenza virus and Mycobacterium tuberculosis. Infect. Immun. 2011, 79, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wu, W.; Zhang, W.; Booth, J.L.; Duggan, E.S.; Tian, L.; More, S.; Zhao, Y.D.; Sawh, R.N.; Liu, L.; et al. RIG-I overexpression decreases mortality of cigarette smoke exposed mice during influenza A virus infection. Respir. Res. 2017, 18, 166. [Google Scholar] [CrossRef]
- Hong, M.J.; Gu, B.H.; Madison, M.C.; Landers, C.; Tung, H.Y.; Kim, M.; Yuan, X.; You, R.; Machado, A.A.; Gilbert, B.E.; et al. Protective role of gammadelta T cells in cigarette smoke and influenza infection. Mucosal. Immunol. 2018, 11, 894–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thatcher, T.H.; McHugh, N.A.; Egan, R.W.; Chapman, R.W.; Hey, J.A.; Turner, C.K.; Redonnet, M.R.; Seweryniak, K.E.; Sime, P.J.; Phipps, R.P. Role of CXCR2 in cigarette smoke-induced lung inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L322–L328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, W.; Marion, C.; Singh, R.; Andrews, N.; Lee, C.G.; Elias, J.A.; Dela Cruz, C.S. Regulation of Retinoic Acid Receptor Beta by Interleukin-15 in the Lung during Cigarette Smoking and Influenza Virus Infection. Am. J. Respir. Cell. Mol. Biol. 2015, 53, 822–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, C.S.; Bauer, C.M.T.; Vujicic, N.; Gaschler, G.J.; Lichty, B.D.; Brown, E.G.; Stampfli, M.R. Cigarette smoke impacts immune inflammatory responses to influenza in mice. Am. J. Respir. Crit. Care Med. 2006, 174, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-J.; Homer, R.J.; Gallo, A.; Lee, C.G.; Crothers, K.; Cho, S.J.; Rochester, C.; Cain, H.; Chupp, G.; Yoon, H.J. IL-18 is induced and IL-18 receptor alpha plays a critical role in the pathogenesis of cigarette smoke-induced pulmonary emphysema and inflammation. J. Immunol. 2007, 178, 1948–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.J.; Lee, C.G.; Lee, J.Y.; Cruz, C.S.D.; Chen, Z.J.; Enelow, R.; Elias, J.A. Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J. Clin. Investig. 2008, 118, 2771–2784. [Google Scholar] [CrossRef] [Green Version]
- Gualano, R.C.; Hansen, M.J.; Vlahos, R.; Jones, E.J.; Park-Jones, A.R.; Deliyannis, G.; Turner, S.J.; Duca, K.A.; Anderson, G.P. Cigarette smoke worsens lung inflammation and impairs resolution of influenza infection in mice. Respir. Res. 2008, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, G.J.; Zavitz, C.C.J.; Bauer, C.M.T.; Skrtic, M.; Lindahl, M.; Robbins, C.; Chen, B.; Stampfli, M.R. Cigarette smoke exposure attenuates cytokine production by mouse alveolar macrophages. Am. J. Respir Cell. Mol. Biol. 2008, 38, 218–226. [Google Scholar] [CrossRef]
- Motz, G.T.; Eppert, B.L.; Wortham, B.W.; Amos-Kroohs, R.M.; Flury, J.L.; Wesselkamper, S.C.; Borchers, M.T. Chronic cigarette smoke exposure primes NK cell activation in a mouse model of chronic obstructive pulmonary disease. J. Immunol. 2010, 184, 4460–4469. [Google Scholar] [CrossRef] [Green Version]
- Botelho, F.M.; Bauer, C.M.T.; Finch, D.; Nikota, J.K.; Zavitz, C.C.J.; Kelly, A.; Lambert, K.N.; Piper, S.; Foster, M.L.; Goldring, J.J.; et al. IL-1α/IL-1R1 expression in chronic obstructive pulmonary disease and mechanistic relevance to smoke-induced neutrophilia in mice. PLoS ONE 2011, 6, e28457. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, W.; More, S.; Booth, J.L.; Duggan, E.S.; Liu, L.; Zhao, Y.D.; Metcalf, J.P. Cigarette smoke attenuates the RIG-I-initiated innate antiviral response to influenza infection in two murine models. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L848–L858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yageta, Y.; Ishii, Y.; Morishima, Y.; Ano, S.; Ohtsuka, S.; Matsuyama, M.; Takeuchi, K.; Itoh, K.; Yamamoto, M.; Hizawa, N. Carbocisteine reduces virus-induced pulmonary inflammation in mice exposed to cigarette smoke. Am. J. Respir. Cell. Mol. Biol. 2014, 50, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Wortham, B.W.; Eppert, B.L.; Motz, G.T.; Flury, J.L.; Orozco-Levi, M.; Hoebe, K.; Panos, R.J.; Maxfield, M.; Glasser, S.W.; Senft, A.P. NKG2D mediates NK cell hyperresponsiveness and influenza-induced pathologies in a mouse model of chronic obstructive pulmonary disease. J. Immunol. 2012, 188, 4468–4475. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Ling, M.T.; Mao, H.; Zheng, J.; Liu, M.; Lam, K.T.; Liu, Y.; Tu, W.; Lau, Y.L. Influenza virus-induced lung inflammation was modulated by cigarette smoke exposure in mice. PLoS ONE 2014, 9, e86166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearley, J.; Silver, J.S.; Sanden, C.; Liu, Z.; Berlin, A.A.; White, N.; Mori, M.; Pham, T.H.; Ward, C.K.; Criner, G.J.; et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 2015, 42, 566–579. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Q.; Xie, J.; Xu, Y. Cigarette smoke inhibits BAFF expression and mucosal immunoglobulin A responses in the lung during influenza virus infection. Respir. Res. 2015, 16, 37. [Google Scholar] [CrossRef] [Green Version]
- Bucher, H.; Duechs, M.J.; Tilp, C.; Jung, B.; Erb, K.J. Tiotropium Attenuates Virus-Induced Pulmonary Inflammation in Cigarette Smoke-Exposed Mice. J. Pharmacol. Exp. Ther. 2016, 357, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Mebratu, Y.A.; Smith, K.R.; Agga, G.; Tesfaigzi, Y. Inflammation and emphysema in cigarette smoke-exposed mice when instilled with poly (I:C) or infected with influenza A or respiratory syncytial viruses. Respir. Res. 2016, 17, 75. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.W.; Sharma, L.; Kang, Y.A.; Kim, S.-H.; Chandrasekharan, S.; Losier, A.; Brady, V.; Bermejo, S.; Andrews, N.; Yoon, C.M.; et al. Impact of Cigarette Smoke Exposure on the Lung Fibroblastic Response after Influenza Pneumonia. Am. J. Respir. Cell. Mol. Biol. 2018, 59, 770–781. [Google Scholar] [CrossRef]
- Wu, W.; Tian, L.; Zhang, W.; Booth, J.L.; Ainsua-Enrich, E.; Kovats, S.; Brown, B.R.; Metcalf, J.P. Long-term cigarette smoke exposure dysregulates pulmonary T cell response and IFN-γ protection to influenza virus in mouse. Respir. Res. 2021, 22, 112. [Google Scholar] [CrossRef]
- Ferrero, M.; Garcia, C.; de Almeida, M.D.; da Silva, J.T.B.; Insuela, D.B.R.; Ferreira, T.T.; de Sá Coutinho, D.; Trindade de Azevedo, C.; Machado Rodrigues, E.; Silva, P.; et al. CCR5 Antagonist Maraviroc Inhibits Acute Exacerbation of Lung Inflammation Triggered by Influenza Virus in Cigarette Smoke-Exposed Mice. Pharmaceuticals 2021, 14, 620. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Todd, I.; Fairclough, L.C. The role of CD8 + T lymphocytes in chronic obstructive pulmonary disease: A systematic review. Inflamm. Res. 2021, 70, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tian, L.; Zhang, W.; Booth, J.L.; Ritchey, J.W.; Wu, S.; Xu, C.; Brown, B.R.; Metcalf, J.P. Early IFN-β administration protects cigarette smoke exposed mice against lethal influenza virus infection without increasing lung inflammation. Sci. Rep. 2022, 12, 4080. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Lead Author/Year | Smoking Length | Stimulation | Collection Time | CS Effects | Note |
|---|---|---|---|---|---|
| Thatcher 2005 [72] | 1 h, twice per day for 3 days | none | N/A | inflammation↑, neutrophils ↑, neutrophil chemotactic chemokines MIP-2 and KC ↑ | |
| Robbins 2006 [74] | 2 cigarettes/d, 5 d/wk, for 3–5 months | low dose or high dose IAV H1N1 (A/FM/1/47) | Day 3, 5, and 7 after infection | inflammation with low-dose infection ↓, inflammation with high-dose influenza ↑ | |
| Kang 2007 [75] | twice per day, 5 d/wk. for 2 wks, 1 month, or 2 months | none | N/A | IL-18, caspases 1 and 11 ↑, inflammation and emphysema ↑ | |
| Kang, 2008 [76] | 3 cigarettes/d for 2 weeks | poly (I:C), IAV H1N1 (A/PR8/34) | Day 3, 9, and 15 after infection | pulmonary inflammation and injury ↑ production of IL-18, IL-12/ IL-23p40, IFN-γ, and type I IFNs ↑ | |
| Gualano 2008 [77] | 9 cigarettes/d for 4 days | IAV H3N1 (Mem71) | Day 3 and 10 after infection | virus titers ↑ macrophages, neutrophils and total lymphocytes ↑ | |
| Gaschler 2008 [78] | twice daily, 5 d/wk for 8 weeks | ex vivo stimulation poly (I:C) and LPS CpG | 2, 6, 24 h after stimulation | TNF-α, IL-6 and RANTES ↓, nuclear translocation NF-κB ↓, AP-1 ↑ | |
| Motz 2010 [79] | 4 h/d, 5 d/wk, for 2, 8 24 weeks | ex vivo stimulation poly (I:C), ssRNA40, or ODN1826 (TLR9 agonist) | 20 h after stimulation | NK cell-derived IFN-γ ↑ | No difference at 2 wks; the difference emerged after 8 wks of CS exposure |
| Feng 2011 [69] | 2h, twice daily, 5 d/wk for 6 weeks | IAV A/PR8/34 | Day 7 after infection | weight loss ↑, pulmonary T-cell response ↓ | |
| Botelho 2011 [80] | 50 min, twice daily, either 4 days or 8 weeks | none | N/A | IL-1R1-dependent neutrophilia↑ | |
| Wu 2014 [81] | 2h, twice daily, 5 d/wk for 6 weeks | IAV A/PR8/34 | Day 7 after infection | lung inflammation by CS alone↑ RIG-I, IFNs and IP-10 ↓ IL-6 and TNF-α ↑ | |
| Yageta 2014 [82] | 10 cigarettes daily for 4 days | IAV A/PR8/34 | Day 7 after infection | pulmonary inflammation and injury ↑ | |
| Wortham 2012 [83] | 4 h/d, 5 d/wk for 6 months | IAV H3N2 (HKx31) | Day 4 after infection | NK cell hyperresponsiveness ↑ pulmonary inflammation ↑ viral clearance = | |
| Han 2014 [84] | 2 h per episode, 2 episodes/d for 21 days | IAV H1N1 (pdmH1N1) and H9N2 (H9N2/G1) | Day 1, 3, and 5 after infection | lung inflammation by CS alone ↑. With IAV, inflammatory cytokines and chemokines ↓, macrophages ↓, neutrophils ↓, T cell infiltration ↓and lung damage ↓ | |
| Kearley 2015 [85] | twice daily for 4 days (most data shown) or 5 d/wk for 8 and 16 weeks | IAV H1N1 (A/FM/1/47-MA) | Day 7 and 11 after infection | IFN-α, IL-6, TNF-α, IFN-γ↑ via IL-33 ↑ | |
| Wang 2015 [86] | 1 cigarette twice/d smoked for 1, 3 and 5 months. | IAV A/PR8/34 | Day 1, 7, and 14 after infection | KC mRNA highest at 1 month after CS alone. KC protein highest at Day 7 after infection. IgA responses ↓ | |
| Wang 2015 [71] | 1 cigarette twice/d smoked for 1 (most data shown), 3 and 6 months | IAV A/PR8/34 | Day 7 after infection | retinoic acid (RA) signaling ↓ | |
| Bucher 2016 [87] | 4 cigarettes/d for 10 days | IAV A/PR8/34 | Day 5 after infection | total cells in BALF Macrophage, neutrophil ↓ KC, IL-6, TNF-α, IL-1β, IFN-γ ↑ | |
| Mebratu 2016 [88] | 6 h/d,5 days/wk, for 4 weeks | IAV HKx31 RSV, poly(I:C) | Day 14 after infection | inflammation was characterized by macrophages, lymphocytes, and neutrophils ↑ | |
| Wang 2017 [70] | 2 h, twice daily, 5 d/wk for 6 weeks | IAV A/PR8/34 | Day 2, 4, and 6 after infection | weight loss ↑ lung inflammation by IAV =, RIG-I, IFN-β and IP-10 ↓ IL-6 and TNF-α before day 4 ↓, Day 6 = | |
| Hong, 2017 [71] | 4 cigarettes/d, 5 d/wk, for 3 months | IAV H3N2 (A/Hong Kong/8/68) | Day 15 after infection | IL-17A, TNF-α, IL-6, and KC ↑, type I/II IFNs, Granzyme b, Ccl3, MIP-1α MIP-1β, and RANTES ↓ | |
| Lee 2018 [89] | 3 cigarettes/d, for 2 wks + 30 days | IAV A/PR8/34 | Until Day 30 after infection | weight loss↑ neutrophils ↑ lung fibrosis↑ WBC and lymphocytes before Day 9 =, after Day 9 ↑ macrophages before Day 9 ↓, after Day 9 ↑ | There is a difference before and after Day 9 |
| Danov 2020 [48] | 6 cigarettes/d for 3 days, followed by 24 cigarettes/d for the remaining 21 days | poly(I:C) and Ex vivo stimulation with IAV H1N1 (pdmH1N1) | Unknown | inflammatory DCs ↑ disrupted epithelial barrier functions↑, antiviral immune response↓ | |
| Wu 2021 [90] | 2 h, twice daily, 5 d/wk for 6 weeks | IAV A/PR8/34 | Day 7 and 10 after infection | IAV-specific T cell, IFN-γ and total protein in BALF at Day 7↓, at Day 10 ↑ | There is an opposite effect of CS on T cell responses to IAV at Day 7 and 10 after infection |
| Ferrero 2021 [91] | 12 cigarettes daily for 12 days | IAV A/PR8/34 | Day 5 after infection | airway obstruction, neutrophil infiltration ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.; Alexander, J.S.; Metcalf, J.P. In Vivo and In Vitro Studies of Cigarette Smoke Effects on Innate Responses to Influenza Virus: A Matter of Models? Viruses 2022, 14, 1824. https://doi.org/10.3390/v14081824
Wu W, Alexander JS, Metcalf JP. In Vivo and In Vitro Studies of Cigarette Smoke Effects on Innate Responses to Influenza Virus: A Matter of Models? Viruses. 2022; 14(8):1824. https://doi.org/10.3390/v14081824
Chicago/Turabian StyleWu, Wenxin, Jeremy S. Alexander, and Jordan P. Metcalf. 2022. "In Vivo and In Vitro Studies of Cigarette Smoke Effects on Innate Responses to Influenza Virus: A Matter of Models?" Viruses 14, no. 8: 1824. https://doi.org/10.3390/v14081824
APA StyleWu, W., Alexander, J. S., & Metcalf, J. P. (2022). In Vivo and In Vitro Studies of Cigarette Smoke Effects on Innate Responses to Influenza Virus: A Matter of Models? Viruses, 14(8), 1824. https://doi.org/10.3390/v14081824