Assessment of Rapid MinION Nanopore DNA Virus Meta-Genomics Using Calves Experimentally Infected with Bovine Herpes Virus-1

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bovine Foetal Lung Cells Infected with BoHV-1
2.2. Experimental Calves
2.3. Nasal Swabs from Experimental Calves
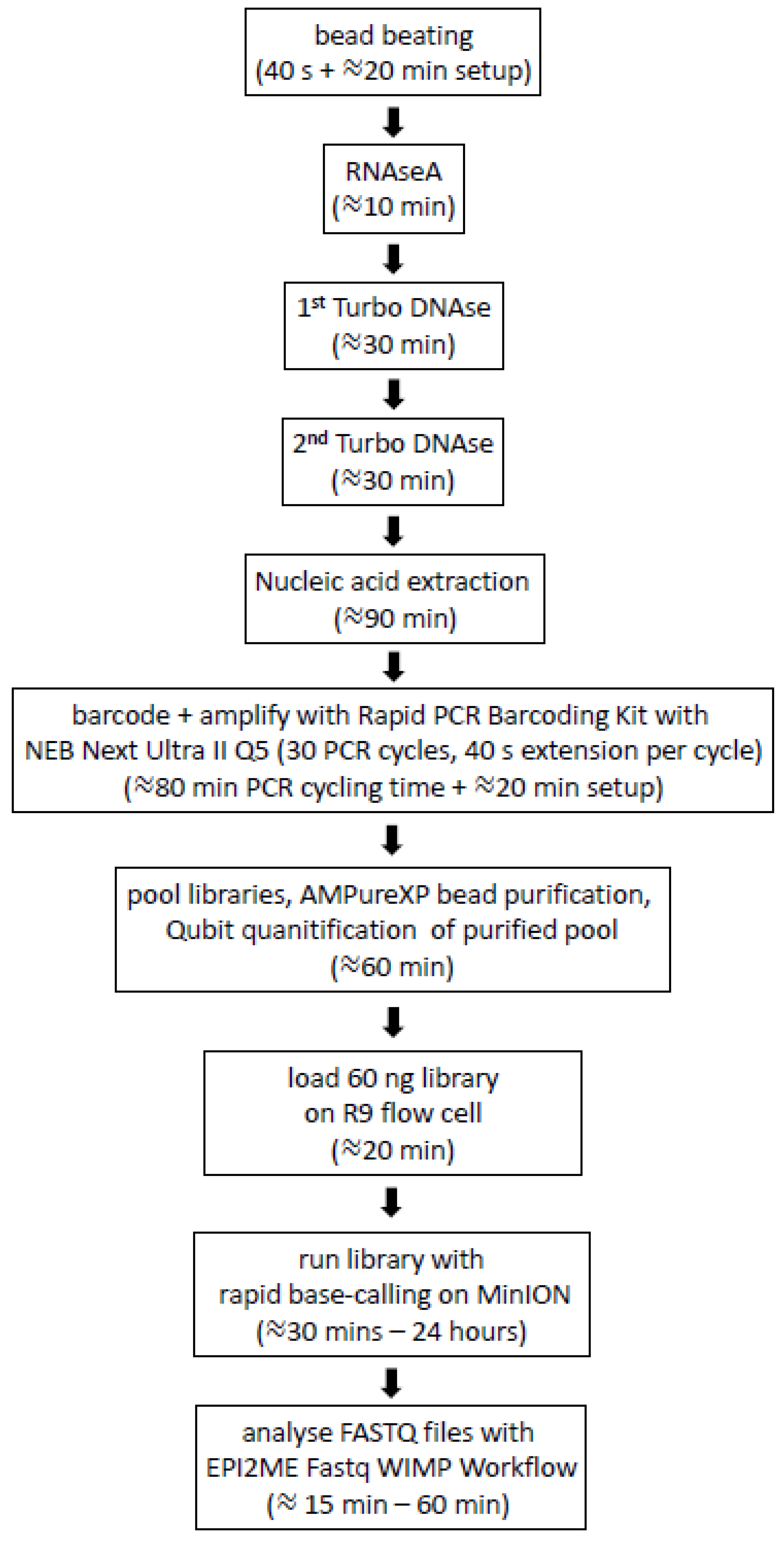
2.4. Non-Viral Nucleic Acid Depletion
2.5. Nucleic Acid Extraction and Purification
2.6. qPCR Analysis
2.7. Generation of MinION Nanopore Libraries for Multiplex Rapid Sequencing
2.8. Epi2ME Analysis
2.9. Sequencing PCR-Free Libraries with the Field Sequencing Kit
2.10. MinION Sequencing of Nasal Swabs Calves Challenged with BoHV-1
2.11. Genome Assemblies
3. Results
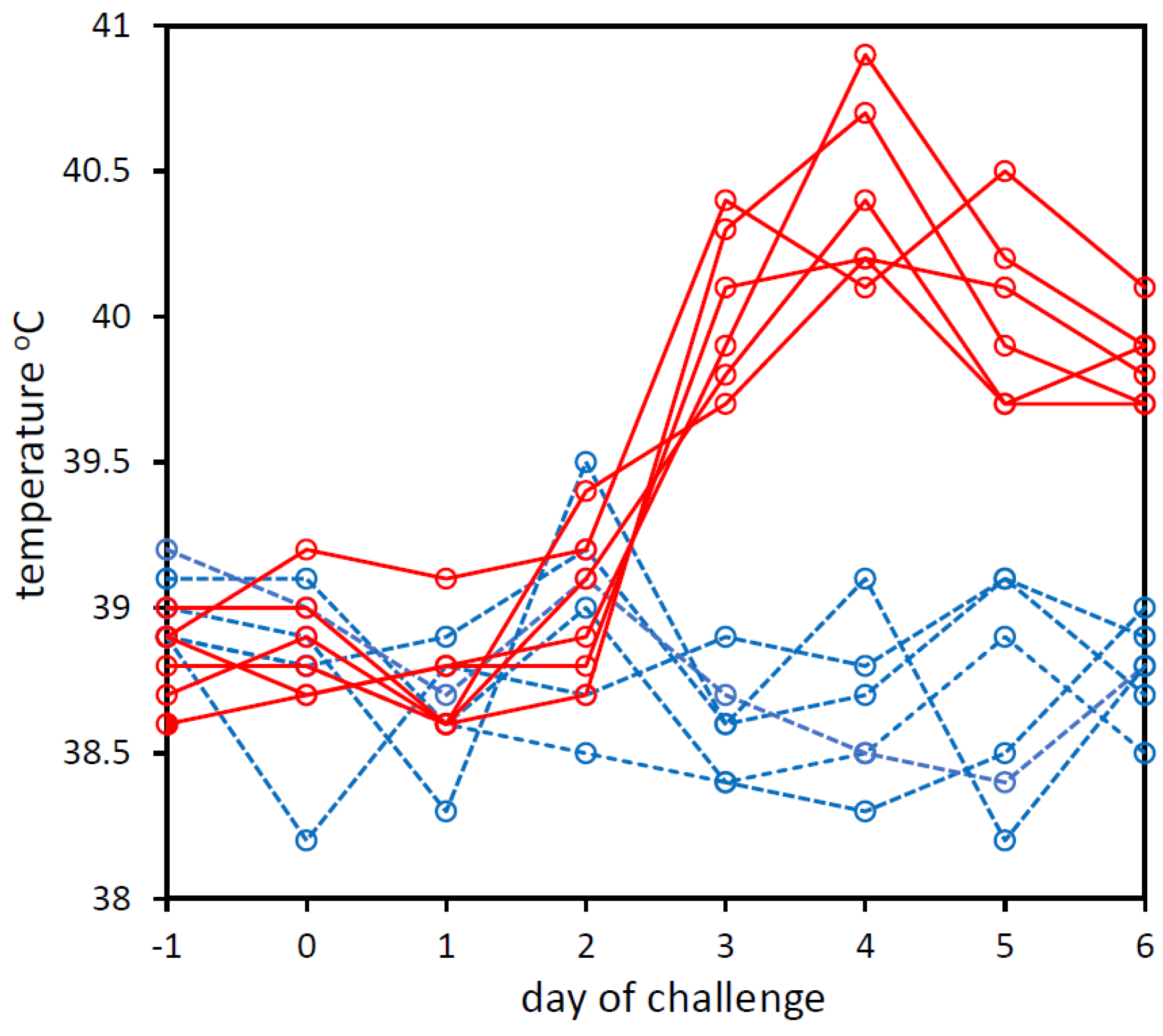
3.1. Experimental Challenge of Calves with BoHV-1 and PBS
3.2. qPCR Analysis of Bovine Nucleic Acid Depletion in BoHV-1-Infected bFLC Cultures
3.3. Effect of Non-Viral Nucleic Acid Depletion on MinION Nanopore Sequencing
3.4. Optimisation of Low-Bias PCR Amplification of Tagmented Libraries
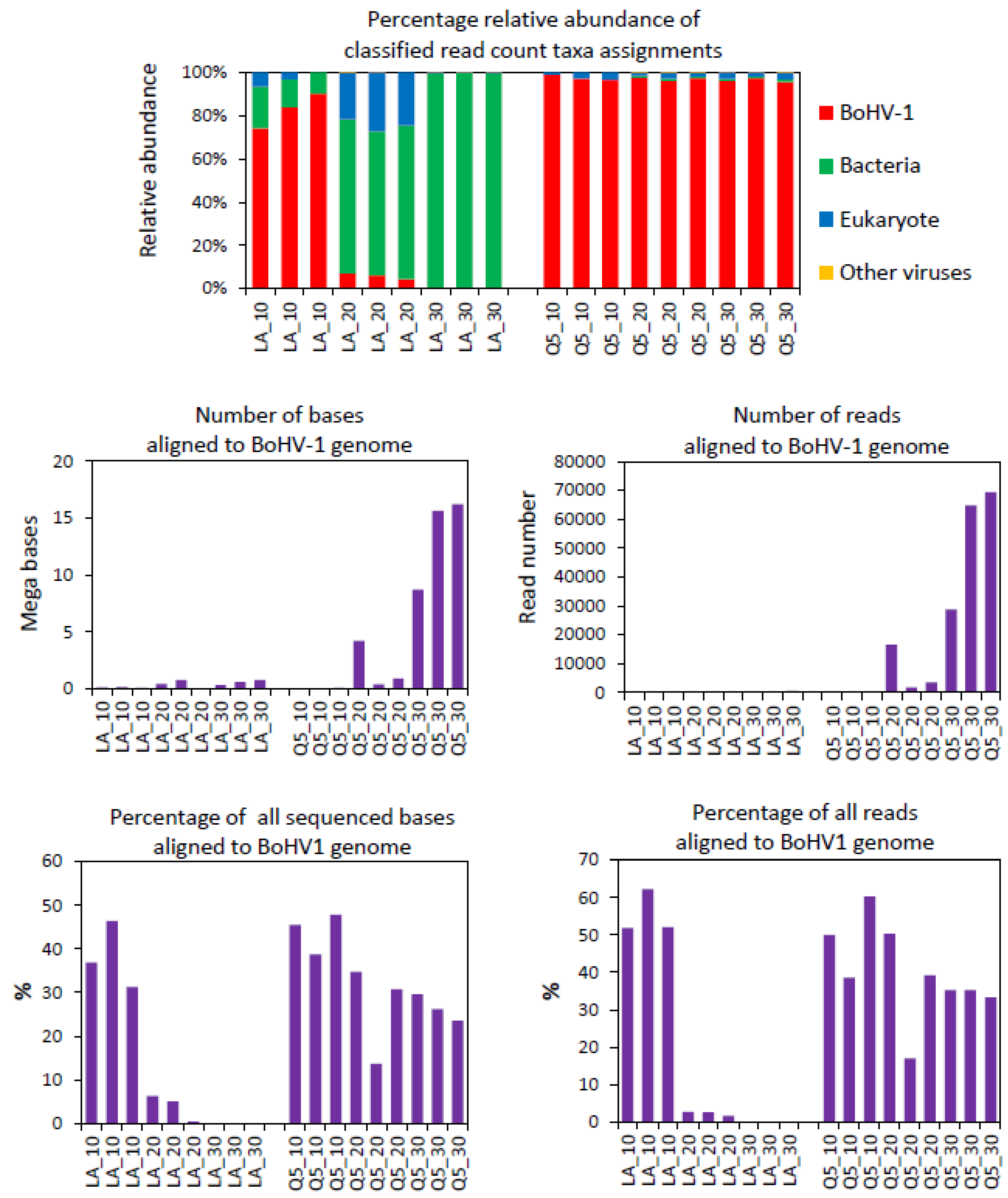
3.5. MinION Sequencing of Nasal Swabs from Calves Challenged with BoHV-1 or PBS
3.6. BoHV-1 Sequence Yield Barcode Variation
3.7. Relationship between Sequencing and qPCR
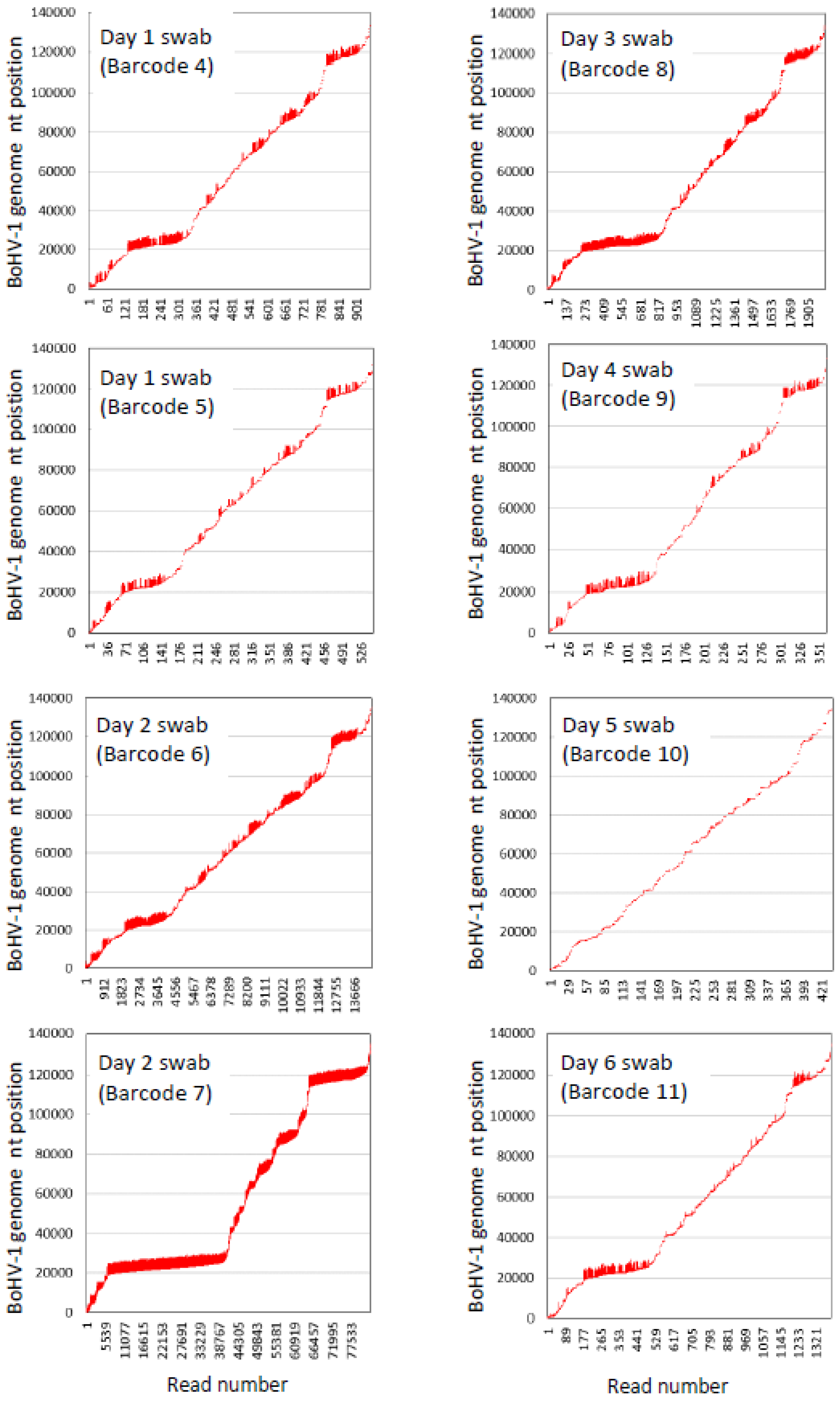
3.8. BoHV-1 Sequence Assembly Direct from Swabs
3.9. Detection of Viruses Other Than BoHV-1 in Nasal Swabs from BoHV-1 Calf Challenge Model
3.10. Incorrect Assignment of Bos taurus Sequence to Viral and Bacterial Taxa by Epi2ME Fastq WIMP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fulton, R.W. Viruses in bovine respiratory disease in North America: Knowledge advances using genomic testing. Vet. Clin. N. Am.-Food Anim. Pract. 2020, 36, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Gómez, I.; McGee, M.; McCabe, M.; Cormican, P.; O’Riordan, E.; McDaneld, T.; Earley, B. Growth performance and hematological changes of weaned beef calves diagnosed with respiratory disease using respiratory scoring and thoracic ultrasonography. J. Anim. Sci. 2020, 98, skaa345. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Gómez, I.; McGee, M.; Sánchez, J.M.; O’Riordan, E.; Byrne, N.; McDaneld, T.; Earley, B. Association between clinical respiratory signs, lung lesions detected by thoracic ultrasonography and growth performance in pre-weaned dairy calves. Ir. Vet. J. 2021, 74, 7. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.; Earley, B.; McCabe, M.S.; Kim, J.; Taylor, J.F.; Lemon, K.; Duffy, C.; McMenamy, M.; Cosby, S.L.; Waters, S.M. Messenger RNA biomarkers of bovine respiratory syncytial virus infection in the whole blood of dairy calves. Sci. Rep. 2021, 11, 9392. [Google Scholar] [CrossRef]
- Johnston, D.; Kim, J.; Taylor, J.F.; Earley, B.; McCabe, M.S.; Lemon, K.; Duffy, C.; McMenamy, M.; Cosby, S.L.; Waters, S.M. ATAC-Seq identifies regions of open chromatin in the bronchial lymph nodes of dairy calves experimentally challenged with bovine respiratory syncytial virus. BMC Genom. 2021, 22, 14. [Google Scholar] [CrossRef]
- Earley, B.; Arguello, A.; O’Riordan, E.; Crosson, P.; McGee, M. Quantifying antimicrobial drug usage from birth to 6 months of age in artificially reared dairy calves and in suckler beef calves. J. Appl. Anim. Res. 2019, 47, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Hope, K.J.; Apley, M.D.; Schrag, N.F.D.; Lubbers, B.V.; Singer, R.S. Antimicrobial use in 22 U.S. beef feed yards: 2016–2017. Zoonoses Public Health 2020, 67 (Suppl. 1), 94–110. [Google Scholar] [CrossRef]
- Ng, T.F.F.; Kondov, N.O.; Deng, X.; Van Eenennaam, A.; Neibergs, H.L.; Delwart, E. A metagenomics and case-control study to identify viruses associated with bovine respiratory disease. J. Virol. 2015, 89, 5340–5349. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Hill, J.E.; Godson, D.L.; Ngeleka, M.; Fernando, C.; Huang, Y. The pulmonary virome, bacteriological and histopathological findings in bovine respiratory disease from western Canada. Transbound. Emerg. Dis. 2019, 67, 924–934. [Google Scholar] [CrossRef]
- Murray, G.M.; More, S.; Sammin, D.; Casey, M.J.; McElroy, M.C.; O’Neill, R.G.; Byrne, W.J.; Earley, B.; Clegg, T.; Ball, H.; et al. Pathogens, patterns of pneumonia, and epidemiologic risk factors associated with respiratory disease in recently weaned cattle in Ireland. J. Vet. Diagn. Investig. 2017, 29, 20–34. [Google Scholar] [CrossRef]
- Dane, H.; Duffy, C.; Guelbenzu, M.; Hause, B.; Fee, S.; Forster, F.; McMenamy, M.J.; Lemon, K. Detection of influenza D virus in bovine respiratory disease samples, UK. Transbound. Emerg. Dis. 2019, 66, 2184–2187. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.; Earley, B.; Cormican, P.; Murray, G.; Kenny, D.A.; Waters, S.M.; McGee, M.; Kelly, A.K.; McCabe, M.S. Illumina MiSeq 16S amplicon sequence analysis of bovine respiratory disease associated bacteria in lung and mediastinal lymph node tissue. BMC Vet. Res. 2017, 13, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosier, D. Review of BRD pathogenesis: The old and the new. Anim. Health Res. Rev. 2014, 15, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Timsit, E.; Dendukuri, N.; Schiller, I.; Buczinski, S. Diagnostic accuracy of clinical illness for bovine respiratory disease (BRD) diagnosis in beef cattle placed in feedlots: A systematic literature review and hierarchical Bayesian latent-class meta-analysis. Prev. Vet. Med. 2016, 135, 67–73. [Google Scholar] [CrossRef]
- Buczinski, S.; Forte, G.; Francoz, D.; Bélanger, A.-M. Comparison of thoracic auscultation, clinical score, and ultrasonography as indicators of bovine respiratory disease in preweaned dairy Calves. J. Vet. Intern. Med. 2014, 28, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Pansri, P.; Katholm, J.; Krogh, K.; Aagaard, A.; Schmidt, L.; Kudirkiene, E.; Larsen, L.E.; Olsen, J. Evaluation of novel multiplex qPCR assays for diagnosis of pathogens associated with the bovine respiratory disease complex. Vet. J. 2020, 256, 105425. [Google Scholar] [CrossRef]
- Kishimoto, M.; Tsuchiaka, S.; Rahpaya, S.S.; Hasebe, A.; Otsu, K.; Sugimura, S.; Kobayashi, S.; Komatsu, N.; Nagai, M.; Omatsu, T.; et al. Development of a one-run real-time PCR detection system for pathogens associated with bovine respiratory disease complex. J. Vet. Med. Sci. 2017, 79, 517–523. [Google Scholar] [CrossRef] [Green Version]
- Veterinary Laboratory Service and Agri-Food & Biosciences Institute. All-Island Animal Disease Surveillance Report 2019; Veterinary Laboratory Service and Agri-Food & Biosciences Institute: Belfast, UK, 2019; pp. 20–28. [Google Scholar]
- Zhang, M.; Hill, J.E.; Alexander, T.W.; Huang, Y. The nasal viromes of cattle on arrival at western Canadian feedlots and their relationship to development of bovine respiratory disease. Transbound. Emerg. Dis. 2020, 68, 2209–2218. [Google Scholar] [CrossRef]
- Kaszab, E.; Doszpoly, A.; Lanave, G.; Verma, A.; Bányai, K.; Marton, S. Metagenomics revealing new virus species in farm and pet animals and aquaculture. In Genomics and Biotechnological Advances in Veterinary, Poultry, and Fisheries; Academic Press: Cambridge, MA, USA, 2020; pp. 29–73. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 124. [Google Scholar] [CrossRef] [Green Version]
- Brock, J.; Lange, M.; Guelbenzu-Gonzalo, M.; Meunier, N.; Vaz, A.M.; Tratalos, J.A.; Dittrich, P.; Gunn, M.; More, S.J.; Graham, D.; et al. Epidemiology of age-dependent prevalence of Bovine Herpes Virus Type 1 (BoHV-1) in dairy herds with and without vaccination. Vet. Res. 2020, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://arriveguidelines.org (accessed on 10 May 2022).
- Ambion by Life Technologies TURBO DNA-Free Kit (Cat. Number AM1907) User Guide. Publication Number 1907M Revision G. 2012. Available online: https://tools.thermofisher.com/content/sfs/manuals/cms_055740.pdf (accessed on 10 May 2022).
- QIAamp Ultra Sens Virus Kit Handbook. 2012. Available online: https://www.qiagen.com/us/resources/resourcedetail?id=c3ca1ee1-a6ef-4421-a7ce-5de99d283629&lang=en (accessed on 10 May 2022).
- Oxford Nanopore Technologies Rapid PCR Barcoding Kit (SQK-RPB004) User Guide. 2017, pp. 1–50. Available online: https://store.nanoporetech.com/eu/rapid-pcr-barcoding-kit.html (accessed on 10 May 2022).
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushnell, B. BBTools Software Package. 2014. Available online: http://sourceforge.net/projects/bbmap (accessed on 10 May 2022).
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- GitHub-Nanoporetech/Medaka: Sequence Correction Provided by ONT Research. Available online: https://github.com/nanoporetech/medaka (accessed on 10 May 2022).
- Dabney, J.; Meyer, M. Length and GC-biases during sequencing library amplification: A comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. BioTechniques 2012, 52, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Nemoto, R.; Kodana, M.; Tarumoto, N.; Sakai, J.; Kawamura, T.; Ikebuchi, K.; Mitsutake, K.; Murakami, T.; Maesaki, S.; et al. Rapid and accurate species identification of mitis group Streptococci using the MinION nanopore sequencer. Front. Cell. Infect. Microbiol. 2020, 10, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, K.S.; Shutov, O.; Lapoint, R.; Kimelman, M.; Brister, J.R.; O’Sullivan, C. STAT: A fast, scalable, MinHash-based k-mer tool to assess Sequence Read Archive next-generation sequence submissions. Genome Biol. 2021, 22, 270. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies. Available online: https://store.nanoporetech.com/eu/flow-cell-r9-4-1.html (accessed on 10 May 2022).
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Wylie, T.N.; Wylie, K.M.; Herter, B.N.; Storch, G.A. Enhanced virome sequencing using targeted sequence capture. Genome Res. 2015, 25, 1910–1920. [Google Scholar] [CrossRef] [Green Version]
- Wylie, K.M.; Wylie, T.N.; Buller, R.; Herter, B.; Cannella, M.T.; Storch, G.A. Detection of viruses in clinical samples by use of metagenomic sequencing and targeted sequence capture. J. Clin. Microbiol. 2018, 56, e01123-18. [Google Scholar] [CrossRef] [Green Version]
- Schuele, L.; Cassidy, H.; Lizarazo, E.; Strutzberg-Minder, K.; Schuetze, S.; Loebert, S.; Lambrecht, C.; Harlizius, J.; Friedrich, A.; Peter, S.; et al. Assessment of viral targeted sequence capture using nanopore sequencing directly from clinical samples. Viruses 2020, 12, 1358. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Achari, A.; Federman, S.; Yu, G.; Somasekar, S.; Bártolo, I.; Yagi, S.; Mbala-Kingebeni, P.; Kapetshi, J.; Ahuka-Mundeke, S.; et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat. Microbiol. 2020, 5, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parras-Moltó, M.; Rodríguez-Galet, A.; Suárez-Rodríguez, P.; López-Bueno, A. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 2018, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Marrie, T.J.; Auffray, J.-P.; Raoult, D. Mimivirus in Pneumonia Patients. Emerg. Infect. Dis. 2005, 11, 449–452. [Google Scholar] [CrossRef]
- Goya, S.; Valinotto, L.E.; Tittarelli, E.; Rojo, G.L.; Jodar, M.S.N.; Greninger, A.L.; Zaiat, J.J.; Martí, M.A.; Mistchenko, A.S.; Viegas, M. An optimized methodology for whole genome sequencing of RNA respiratory viruses from nasopharyngeal aspirates. PLoS ONE 2018, 13, e0199714. [Google Scholar] [CrossRef] [Green Version]
- Kallies, R.; Hölzer, M.; Toscan, R.B.; da Rocha, U.N.; Anders, J.; Marz, M.; Chatzinotas, A. Evaluation of sequencing library preparation protocols for viral metagenomic analysis from pristine aquifer groundwaters. Viruses 2019, 11, 484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Huang, Y.; Godson, D.L.; Fernando, C.; Alexander, T.W.; Hill, J.E. Assessment of metagenomic sequencing and qPCR for detection of influenza d virus in bovine respiratory tract samples. Viruses 2020, 12, 814. [Google Scholar] [CrossRef]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smithm, A.D.; et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 2020. [Google Scholar] [CrossRef]
- McCabe, M.S.; Cormican, P.; Johnston, D.; Earley, B. Simultaneous detection of DNA and RNA virus species involved in bovine respiratory disease by PCR-free rapid tagmentation-based library preparation and MinION nanopore sequencing. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Dhufaigh, K.N.; McCabe, M.; Cormican, P.; Cuevas-Gomez, I.; McGee, M.; McDaneld, T.; Earley, B. Nanopore sequencing of the respiratory virome in beef-suckler weanlings diagnosed with bovine respiratory disease. In Proceedings of the Animal—Science Proceedings V British Society of Animal Science’s Annual Conference, Nottingham, UK, 12–14 April 2022; Volume 13, pp. 70–71. [Google Scholar] [CrossRef]
- Wu, X.; Luo, H.; Xu, F.; Ge, C.; Li, S.; Deng, X.; Wiedmann, M.; Baker, R.C.; Stevenson, A.; Zhang, G.; et al. Evaluation of Salmonella serotype prediction with multiplex nanopore sequencing. Front. Microbiol. 2021, 12, 637771. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies Voltrax. Available online: https://nanoporetech.com/products/voltrax (accessed on 10 May 2022).
- MicroGEM PDQeX Nucleic Acid Extractor. Available online: https://microgembio.com/products/virus/ (accessed on 29 July 2022).
- López-Labrador, F.X.; Brown, J.R.; Fischer, N.; Harvala, H.; Van Boheemen, S.; Cinek, O.; Sayiner, A.; Madsen, T.V.; Auvinen, E.; Kufner, V.; et al. Recommendations for the introduction of metagenomic high-throughput sequencing in clinical virology, part I: Wet lab procedure. J. Clin. Virol. 2021, 134, 104691. [Google Scholar] [CrossRef] [PubMed]
- Jurasz, H.; Pawłowski, T.; Perlejewski, K. Contamination Issue in Viral Metagenomics: Problems, Solutions, and Clinical Perspectives. Front. Microbiol. 2021, 12, 745076. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Matthijnssens, J.; Dutilh, B.E. Metagenomics in Virology. Encycl. Virol. 2021, 1, 133–140. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | BoHV-1 Cq (UL27) | s.d. | Bovine Cq (bACTB) | s.d. | BoHV-1 (UL27) Relative Quantities | Bovine (bACTB) Relative Quantities |
|---|---|---|---|---|---|---|
| A | 20.76 | 0.09 | 32.00 | 0.21 | 1.00 | 1.00 |
| B | 24.79 | 0.40 | 35.72 | 0.67 | −16.37 | −13.20 |
| C | 20.60 | 0.54 | 33.80 | 1.75 | 1.11 | −3.48 |
| D | 21.70 | 0.15 | 35.20 | 0.92 | −1.92 | −9.19 |
| E | 24.04 | 0.37 | 38.33 | 1.49 | −9.77 | −80.63 |
| Taxonomic Assignment | Undepleted | Depleted | ||||||
|---|---|---|---|---|---|---|---|---|
| bFLC1 | bFLC2 | bFLC3 | Swab | bFLC1 | bFLC2 | bFLC3 | Swab | |
| Number of Reads Assigned to BoHV-1 | ||||||||
| Eukaryota | >31,850 | 14,427 | 33,459 | 154,002 | 75 | 54 | 51 | 12,541 |
| BoHV-1 | 24,295 | 11,854 | 27,180 | 501 | 4941 | 3339 | 3059 | 1781 |
| Other viruses | 1868 | 660 | 2022 | 156 | 492 | 338 | 325 | 122 |
| Bacteria | 528 | 252 | 563 | 2057 | 90 | 62 | 60 | 279 |
| Archaea | 31 | 11 | 31 | 126 | 0 | 0 | 0 | 13 |
| Classified | 58,759 | 27,289 | 63,480 | 157,738 | 5604 | 3797 | 3502 | 14,799 |
| Unclassified | 246,018 | 75,995 | 253,937 | 341,679 | 7555 | 5794 | 5280 | 229,354 |
| Percentage of classified reads assigned to BoHV-1 | ||||||||
| Eukaryota | 54.38 | 53.03 | 52.90 | 98.19 | 1.34 | 1.42 | 1.46 | 85.10 |
| BoHV-1 | 41.35 | 43.44 | 42.82 | 0.32 | 88.17 | 87.94 | 87.35 | 12.03 |
| Other viruses | 3.18 | 2.42 | 3.19 | 0.10 | 8.78 | 8.90 | 9.28 | 0.82 |
| Bacteria | 0.90 | 0.93 | 0.89 | 1.31 | 1.61 | 1.63 | 1.72 | 1.89 |
| Archaea | 0.05 | 0.04 | 0.05 | 0.08 | 0.00 | 0.00 | 0.00 | 0.09 |
| Percentage of classified and unclassified reads assigned to BoHV-1 | ||||||||
| Eukaryota | 10.45 | 13.97 | 10.54 | 30.84 | 0.57 | 0.56 | 0.58 | 5.14 |
| BoHV-1 | 7.97 | 11.48 | 8.56 | 0.10 | 37.55 | 34.81 | 34.83 | 0.73 |
| Other viruses | 0.61 | 0.64 | 0.64 | 0.03 | 3.74 | 3.52 | 3.70 | 0.05 |
| Bacteria | 0.21 | 0.33 | 0.22 | 0.60 | 1.19 | 1.07 | 1.14 | 0.12 |
| Archaea | 31.30 | 11.11 | 31.29 | 127.78 | 0.00 | 0.00 | 0.00 | 13.26 |
| Taxonomic Assignment | Sequence Read Counts | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 10 PCR Cycles | 20 PCR Cycles | 30 PCR Cycles | |||||||
| BoHV-1 (LongAmp) | 81 | 115 | 65 | 59 | 116 | 8 | 206 | 342 | 421 |
| BoHV-1 (Q5) | 231 | 36 | 92 | 3299 | 1640 | 15,179 | 69,738 | 65,305 | 28,423 |
| Bacteria (LongAmp) | 21 | 18 | 7 | 612 | 1273 | 131 | 414,892 | 1,094,002 | 1,161,715 |
| Bacteria (Q5) | 0 | 0 | 0 | 31 | 19 | 119 | 620 | 505 | 370 |
| Eukaryote (LongAmp) | 7 | 4 | 0 | 183 | 516 | 45 | 1361 | 2710 | 3451 |
| Eukaryote (Q5) | 2 | 1 | 3 | 43 | 43 | 254 | 2052 | 1119 | 877 |
| Other viruses (LongAmp) | 0 | 0 | 0 | 1 | 1 | 0 | 163 | 211 | 282 |
| Other viruses (Q5) | 0 | 0 | 0 | 9 | 2 | 15 | 56 | 54 | 25 |
| Calf No. | Day (d) Relative to Challenge | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| d −1 | d 0 | d 1 | d 2 | d 3 | d 4 | d 5 | d 6 | -ve | |
| Number of Reads Assigned to BoHV-1 | |||||||||
| PBS_1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PBS_2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PBS_3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PBS_4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PBS_5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PBS_6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BoHV1_1 | 0 | 1 | 635 | 44,864 | 1779 | 314 | 356 | 1186 | 1 |
| BoHV1_2 | 1 | 2 | 69,864 | 3024 | 204,042 | 26,519 | 7 | 884 | 2 |
| BoHV1_3 | 0 | 0 | 14 | 1607 | 1 | 220 | 103 | 2218 | 0 |
| BoHV1_4 | 0 | 0 | 174 | 46,381 | 478 | 259 | 284 | 47 | 0 |
| BoHV1_5 | 0 | 0 | 134 | 1405 | 385 | 939 | 86 | 110 | 0 |
| BoHV1_6 | 0 | 0 | 7 | 554 | 751 | 163 | 355 | 2300 | 0 |
| Percentage of classified reads assigned to BoHV-1 | |||||||||
| PBS_1 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PBS_2 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PBS_3 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PBS_4 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PBS_5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PBS_6 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| BoHV1_1 | 0.00 | 0.01 | 45.50 | 93.04 | 18.50 | 32.85 | 36.00 | 18.13 | 1.22 |
| BoHV1_2 | 0.65 | 0.55 | 48.13 | 4.54 | 72.73 | 16.08 | 1.13 | 0.41 | 0.36 |
| BoHV1_3 | 0.00 | 0.00 | 0.01 | 3.25 | 0.39 | 0.60 | 0.33 | 0.77 | 0.00 |
| BoHV1_4 | 0.00 | 0.00 | 1.31 | 35.90 | 1.35 | 0.08 | 0.11 | 0.02 | 0.00 |
| BoHV1_5 | 0.00 | 0.00 | 0.09 | 2.57 | 4.74 | 3.42 | 1.43 | 0.71 | 0.00 |
| BoHV1_6 | 0.00 | 0.00 | 0.00 | 4.70 | 1.40 | 1.83 | 2.66 | 0.50 | 0.00 |
| Percentage of classified and unclassified reads assigned to BoHV-1 | |||||||||
| PBS_1 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PBS_2 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PBS_3 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PBS_4 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PBS_5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PBS_6 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| BoHV1_1 | 0.00 | 0.00 | 8.23 | 71.40 | 1.26 | 1.94 | 0.97 | 6.10 | 0.13 |
| BoHV1_2 | 0.65 | 0.81 | 34.13 | 1.99 | 65.94 | 11.95 | 1.49 | 0.47 | 0.00 |
| BoHV1_3 | 0.00 | 0.00 | 0.01 | 2.54 | 0.24 | 0.37 | 0.17 | 0.41 | 0.00 |
| BoHV1_4 | 0.00 | 0.00 | 0.89 | 34.64 | 0.95 | 0.05 | 0.08 | 0.02 | 0.00 |
| BoHV1_5 | 0.00 | 0.00 | 0.07 | 2.27 | 2.68 | 2.74 | 0.83 | 0.38 | 0.00 |
| BoHV1_6 | 0.00 | 0.00 | 0.00 | 2.64 | 1.03 | 3.35 | 1.66 | 0.42 | 0.00 |
| Day Relative to Challenge | Barcode | Number of Aligned Bases (kb) | Number of Unaligned Bases (kb) | Number of Aligned Reads | Number of Unaligned Reads | Bases That Aligned (%) | Average Length of Aligned Reads | Average Length of Unaligned Reads | Average Identity of BoHV-1-Aligned Reads (%) |
|---|---|---|---|---|---|---|---|---|---|
| −1 | 1 | 0 | 3700 | 0 | 62,15 | 0.0 | 0.0 | 595.3 | 0 |
| 0 | 2 | 0 | 28,000 | 0 | 28,657 | 0.0 | 0.0 | 977.1 | 0 |
| 0 | 3 | 0 | 73,400 | 0 | 39,985 | 0.0 | 0.0 | 1835.7 | 0 |
| 1 | 4 | 1200 | 8200 | 948 | 8832 | 12.8 | 1265.8 | 928.4 | 95.5 |
| 1 | 5 | 546 | 5253.8 | 549 | 5320 | 9.4 | 994.9 | 987.6 | 95.5 |
| 2 | 6 | 12,400 | 6100 | 14,567 | 4617 | 67.0 | 851.2 | 1321.2 | 95.4 |
| 2 | 7 | 223,600 | 37,000 | 83,070 | 17,099 | 85.8 | 2691.7 | 2163.9 | 95.4 |
| 3 | 8 | 3000 | 163,900 | 2032 | 141,366 | 1.8 | 1476.4 | 1159.4 | 95.4 |
| 4 | 9 | 624 | 14,875.1 | 361 | 16,133 | 4.0 | 1731.0 | 922.0 | 95.5 |
| 5 | 10 | 173 | 34,026.7 | 438 | 36,820 | 0.5 | 395.7 | 924.1 | 95.5 |
| 6 | 11 | 1400 | 22,000 | 1397 | 18,351 | 6.0 | 1002.1 | 1198.8 | 95.5 |
| PBS | 12 | 3.1 | 390.3 | 1 | 773 | 0.8 | 3100.0 | 504.9 | 94.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esnault, G.; Earley, B.; Cormican, P.; Waters, S.M.; Lemon, K.; Cosby, S.L.; Lagan, P.; Barry, T.; Reddington, K.; McCabe, M.S. Assessment of Rapid MinION Nanopore DNA Virus Meta-Genomics Using Calves Experimentally Infected with Bovine Herpes Virus-1. Viruses 2022, 14, 1859. https://doi.org/10.3390/v14091859
Esnault G, Earley B, Cormican P, Waters SM, Lemon K, Cosby SL, Lagan P, Barry T, Reddington K, McCabe MS. Assessment of Rapid MinION Nanopore DNA Virus Meta-Genomics Using Calves Experimentally Infected with Bovine Herpes Virus-1. Viruses. 2022; 14(9):1859. https://doi.org/10.3390/v14091859
Chicago/Turabian StyleEsnault, Gaelle, Bernadette Earley, Paul Cormican, Sinead M. Waters, Ken Lemon, S. Louise Cosby, Paula Lagan, Thomas Barry, Kate Reddington, and Matthew S. McCabe. 2022. "Assessment of Rapid MinION Nanopore DNA Virus Meta-Genomics Using Calves Experimentally Infected with Bovine Herpes Virus-1" Viruses 14, no. 9: 1859. https://doi.org/10.3390/v14091859
APA StyleEsnault, G., Earley, B., Cormican, P., Waters, S. M., Lemon, K., Cosby, S. L., Lagan, P., Barry, T., Reddington, K., & McCabe, M. S. (2022). Assessment of Rapid MinION Nanopore DNA Virus Meta-Genomics Using Calves Experimentally Infected with Bovine Herpes Virus-1. Viruses, 14(9), 1859. https://doi.org/10.3390/v14091859