
SARS-CoV-2 Genetic Diversity and Lineage Dynamics in Egypt during the First 18 Months of the Pandemic

 , , , , , , , , , ,
, , , , , , , , , ,  ,
,  ,
,  , and
, and 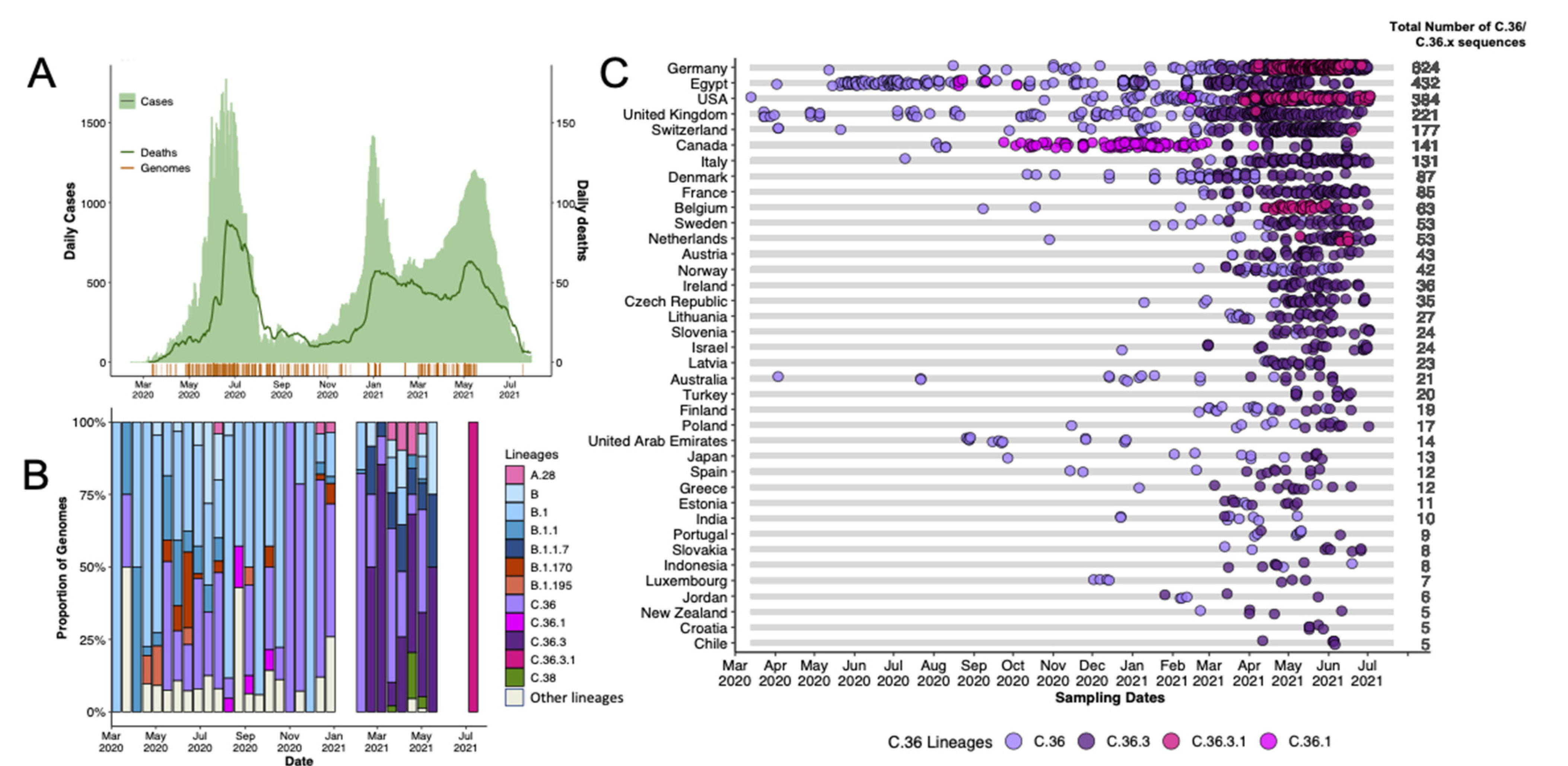{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Epidemiological Data
2.3. qPCR analyzed Samples
2.4. Mutation Screening Assays
2.5. SARS-CoV-2 Whole-Genome Sequencing
2.6. Genomic Analysis and Strain Typing
2.7. Phylogenetic Analysis
3. Results
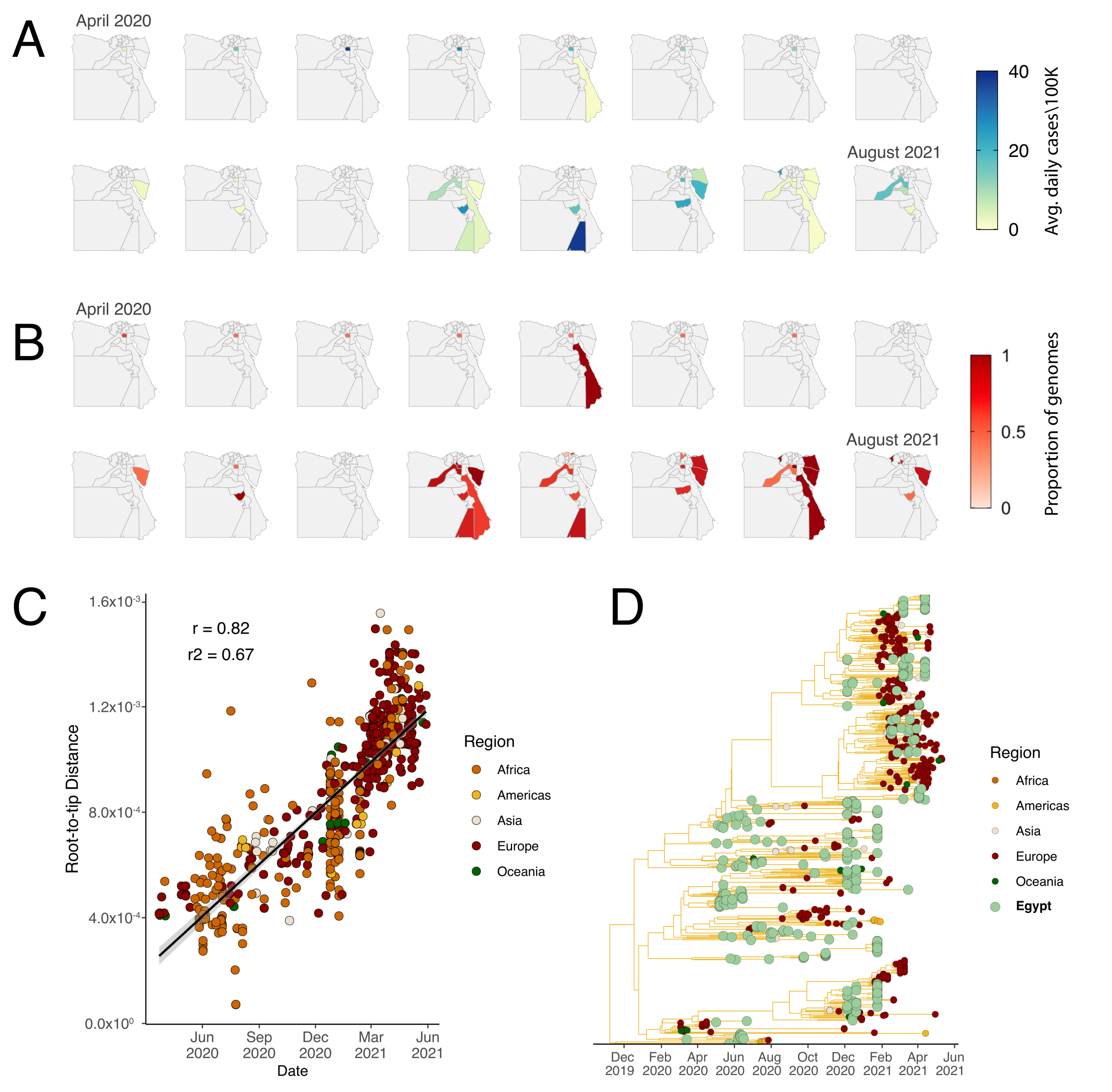
3.1. Genomic Epidemiology of SARS-CoV-2 in Egypt

3.2. Phylogenetic Analysis and Lineage Dynamics

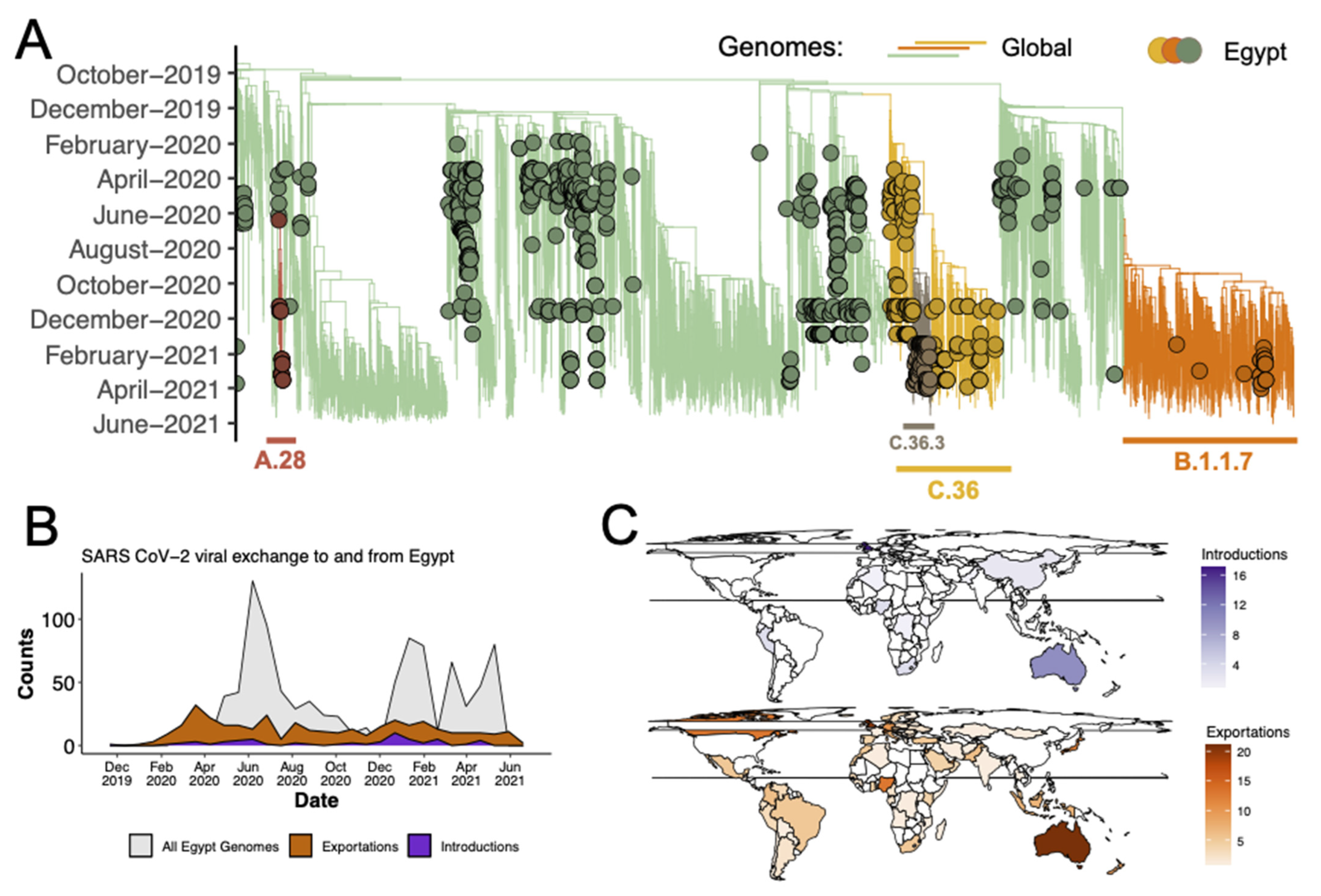
3.3. Introduction of the C.36 lineage in Egypt
3.4. Evolution and Spread of C.36 Sub-Lineages

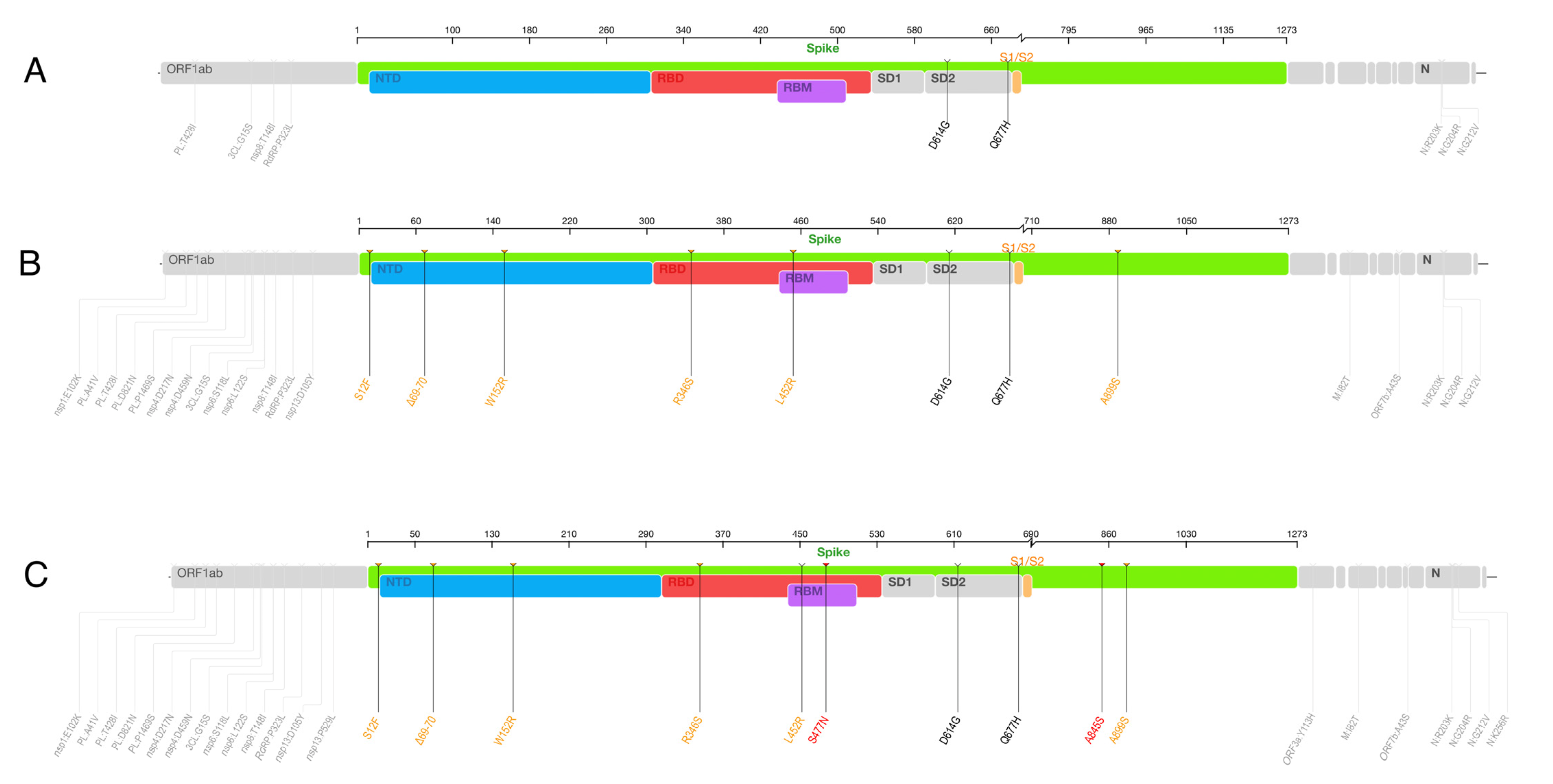
3.5. Mutational and Selection Analysis of the C.36.3 Lineage

4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- ECDC. SARS-CoV-2 Variants of Concern as of 18 November 2021. Available online: https://www.ecdc.europa.eu/en (accessed on 9 August 2021).
- Gomaa, M.R.; El Rifay, A.S.; Shehata, M.; Kandeil, A.; Nabil Kamel, M.; Marouf, M.A.; GabAllah, M.; El Taweel, A.; Kayed, A.E.; Kutkat, O.; et al. Incidence, household transmission, and neutralizing antibody seroprevalence of Coronavirus Disease 2019 in Egypt: Results of a community-based cohort. PLoS Pathog. 2021, 17, e1009413. [Google Scholar] [CrossRef] [PubMed]
- Kandeil, A.; Mostafa, A.; El-Shesheny, R.; Shehata, M.; Roshdy, W.H.; Ahmed, S.S.; Gomaa, M.; Taweel, A.E.; Kayed, A.E.; Mahmoud, S.H.; et al. Coding-Complete Genome Sequences of Two SARS-CoV-2 Isolates from Egypt. Microbiol. Resour. Announc. 2020, 9, e00489-20. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. COVID-19 Weekly Epidemiological Update 2021; World Health Organization: Geneva, Switzerland, 2021.
- Our World in Data, O. COVID-19 Dataset by Our World in Data. Available online: https://github.com/owid/covid-19-data (accessed on 29 September 2021).
- Huisman, J.S.; Scire, J.; Angst, D.C.; Li, J.; Neher, R.A.; Maathuis, M.H.; Bonhoeffer, S.; Stadler, T. Estimation and worldwide monitoring of the effective reproductive number of SARS-CoV-2. medRxiv 2021. [Google Scholar] [CrossRef]
- Huisman, J.S. Daily SARS-CoV-2 Re Values for Select Countries. Available online: https://github.com/covid-19-Re/dailyRe-Data (accessed on 6 October 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A.; Kelso, J. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Neher, R. Nextalign: Viral Genome Reference Alignment. Available online: https://github.com/neherlab/nextalign (accessed on 16 December 2021).
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. Corrigendum to: IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 2461. [Google Scholar] [CrossRef] [PubMed]
- Tavare, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Some Math. Quest. Biol./DNA Seq. Anal. 1986, 17, 57–86. [Google Scholar]
- Lemoine, F.; Domelevo Entfellner, J.B.; Wilkinson, E.; Correia, D.; Davila Felipe, M.; De Oliveira, T.; Gascuel, O. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 2018, 556, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E.; Giovanetti, M.; Tegally, H.; San, J.E.; Lessells, R.; Cuadros, D.; Martin, D.P.; Rasmussen, D.A.; Zekri, A.-R.N.; Sangare, A.K.; et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Science 2021, 374, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y.; et al. The emergence and ongoing convergent evolution of the SARS-CoV-2 N501Y lineages. Cell 2021, 184, 5189–5200. [Google Scholar] [CrossRef] [PubMed]
- Saied, A.A.; Metwally, A.A.; Madkhali, N.A.B.; Haque, S.; Dhama, K. Egypt’s COVID-19 Recent Happenings and Perspectives: A Mini-Review. Front. Public Health 2021, 9, 696082. [Google Scholar] [CrossRef] [PubMed]
- Girgis, S.A.; Hafez, H.M.; Elarab, H.E.; Sherif, B.; Sabry, M.H.; Afifi, I.; Hassan, F.E.; Reda, A.; Elsayed, S.; Mahmoud, A.; et al. SARS-CoV-2 PCR positivity rate and seroprevalence of related antibodies among a sample of patients in Cairo: Pre-wave 2 results of a screening program in a university hospital. PLoS ONE 2021, 16, e0254581. [Google Scholar] [CrossRef] [PubMed]
- El-Sokkary, R.H.; Daef, E.; El-Korashi, L.A.; Khedr, E.M.; Gad, D.; Mohamed-Hussein, A.; Zayed, N.E.; Mostafa, E.F.; Bahgat, S.M.; Hassany, S.M.; et al. Sero-prevalence of anti-SARS-CoV-2 antibodies among healthcare workers: A multicenter study from Egypt. J. Infect. Public Health 2021, 14, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Landier, J.; Paireau, J.; Rebaudet, S.; Legendre, E.; Lehot, L.; Fontanet, A.; Cauchemez, S.; Gaudart, J. Cold and dry winter conditions are associated with greater SARS-CoV-2 transmission at regional level in western countries during the first epidemic wave. Sci. Rep. 2021, 11, 12756. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Domman, D.B.; Snyder, D.J.; Oguntuyo, K.Y.; Van Diest, M.; Densmore, K.H.; Schwalm, K.C.; Femling, J.; Carroll, J.L.; Scott, R.S.; et al. Emergence in late 2020 of multiple lineages of SARS-CoV-2 Spike protein variants affecting amino acid position 677. medRxiv 2021. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amanat, F.; Thapa, M.; Lei, T.; Ahmed, S.M.S.; Adelsberg, D.C.; Carreño, J.M.; Strohmeier, S.; Schmitz, A.J.; Zafar, S.; Zhou, J.Q.; et al. SARS-CoV-2 mRNA vaccination induces functionally diverse antibodies to NTD, RBD, and S2. Cell 2021, 184, 3936–3948.e3910. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef] [PubMed]
- Public Health England. SARS-CoV-2 Variants of Concern and Variants under Investigation in England; 2021. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1009243/Technical_Briefing_20.pdf (accessed on 9 August 2021).
- Timmers, L.F.S.M.; Peixoto, J.V.; Ducati, R.G.; Bachega, J.F.R.; de Mattos Pereira, L.; Caceres, R.A.; Majolo, F.; da Silva, G.L.; Anton, D.B.; Dellagostin, O.A.; et al. SARS-CoV-2 mutations in Brazil: From genomics to putative clinical conditions. Sci. Rep. 2021, 11, 11998. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.-Y.; Zhang, Y.; Zhou, X.; Huang, K.; Qian, Y.; Leng, Y.; Yan, L.; Huang, B.; He, Y. Molecular Evolution of SARS-CoV-2 Structural Genes: Evidence of Positive Selection in Spike Glycoprotein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nelson-Sathi, S.; Umasankar, P.K.; Sreekumar, E.; Radhakrishnan Nair, R.; Joseph, I.; Nori, S.R.C.; Philip, J.S.; Prasad, R.; Navyasree, K.V.; Ramesh, S.; et al. Mutational landscape and in silico structure models of SARS-CoV-2 Spike Receptor Binding Domain reveal key molecular determinants for virus-host interaction. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Kubik, S.; Arrigo, N.; Bonet, J.; Xu, Z. Mutational Hotspot in the SARS-CoV-2 Spike Protein N-Terminal Domain Conferring Immune Escape Potential. Viruses 2021, 13, 2114. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e1220. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- El-Saadani, S.; Saleh, M.; Ibrahim, S.A. Quantifying non-communicable diseases’ burden in Egypt using State-Space model. PLoS ONE 2021, 16, e0245642. [Google Scholar] [CrossRef]
- Weigang, S.; Fuchs, J.; Zimmer, G.; Schnepf, D.; Kern, L.; Beer, J.; Luxenburger, H.; Ankerhold, J.; Falcone, V.; Kemming, J.; et al. Within-host evolution of SARS-CoV-2 in an immunosuppressed COVID-19 patient as a source of immune escape variants. Nat. Commun. 2021, 12, 6405. [Google Scholar] [CrossRef]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 2021, 184, 5179–5188.e5178. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Lessells, R.J.; Giandhari, J.; Wolter, N.; Everatt, J.; Rambaut, A.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. medRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roshdy, W.H.; Khalifa, M.K.; San, J.E.; Tegally, H.; Wilkinson, E.; Showky, S.; Martin, D.P.; Moir, M.; Naguib, A.; Elguindy, N.; et al. SARS-CoV-2 Genetic Diversity and Lineage Dynamics in Egypt during the First 18 Months of the Pandemic. Viruses 2022, 14, 1878. https://doi.org/10.3390/v14091878
Roshdy WH, Khalifa MK, San JE, Tegally H, Wilkinson E, Showky S, Martin DP, Moir M, Naguib A, Elguindy N, et al. SARS-CoV-2 Genetic Diversity and Lineage Dynamics in Egypt during the First 18 Months of the Pandemic. Viruses. 2022; 14(9):1878. https://doi.org/10.3390/v14091878
Chicago/Turabian StyleRoshdy, Wael H., Mohamed K. Khalifa, James Emmanuel San, Houriiyah Tegally, Eduan Wilkinson, Shymaa Showky, Darren Patrick Martin, Monika Moir, Amel Naguib, Nancy Elguindy, and et al. 2022. "SARS-CoV-2 Genetic Diversity and Lineage Dynamics in Egypt during the First 18 Months of the Pandemic" Viruses 14, no. 9: 1878. https://doi.org/10.3390/v14091878
APA StyleRoshdy, W. H., Khalifa, M. K., San, J. E., Tegally, H., Wilkinson, E., Showky, S., Martin, D. P., Moir, M., Naguib, A., Elguindy, N., Gomaa, M. R., Fahim, M., Abu Elsood, H., Mohsen, A., Galal, R., Hassany, M., Lessells, R. J., Al-Karmalawy, A. A., EL-Shesheny, R., ... de Oliveira, T. (2022). SARS-CoV-2 Genetic Diversity and Lineage Dynamics in Egypt during the First 18 Months of the Pandemic. Viruses, 14(9), 1878. https://doi.org/10.3390/v14091878