

The Spread of SARS-CoV-2 Omicron Variant in CALABRIA: A Spatio-Temporal Report of Viral Genome Evolution

 , , , , , , ,
, , , , , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Sequencing of Viral Genome
2.3. Phylogenetic Analysis
2.4. Analysis of Viral Strains
3. Results
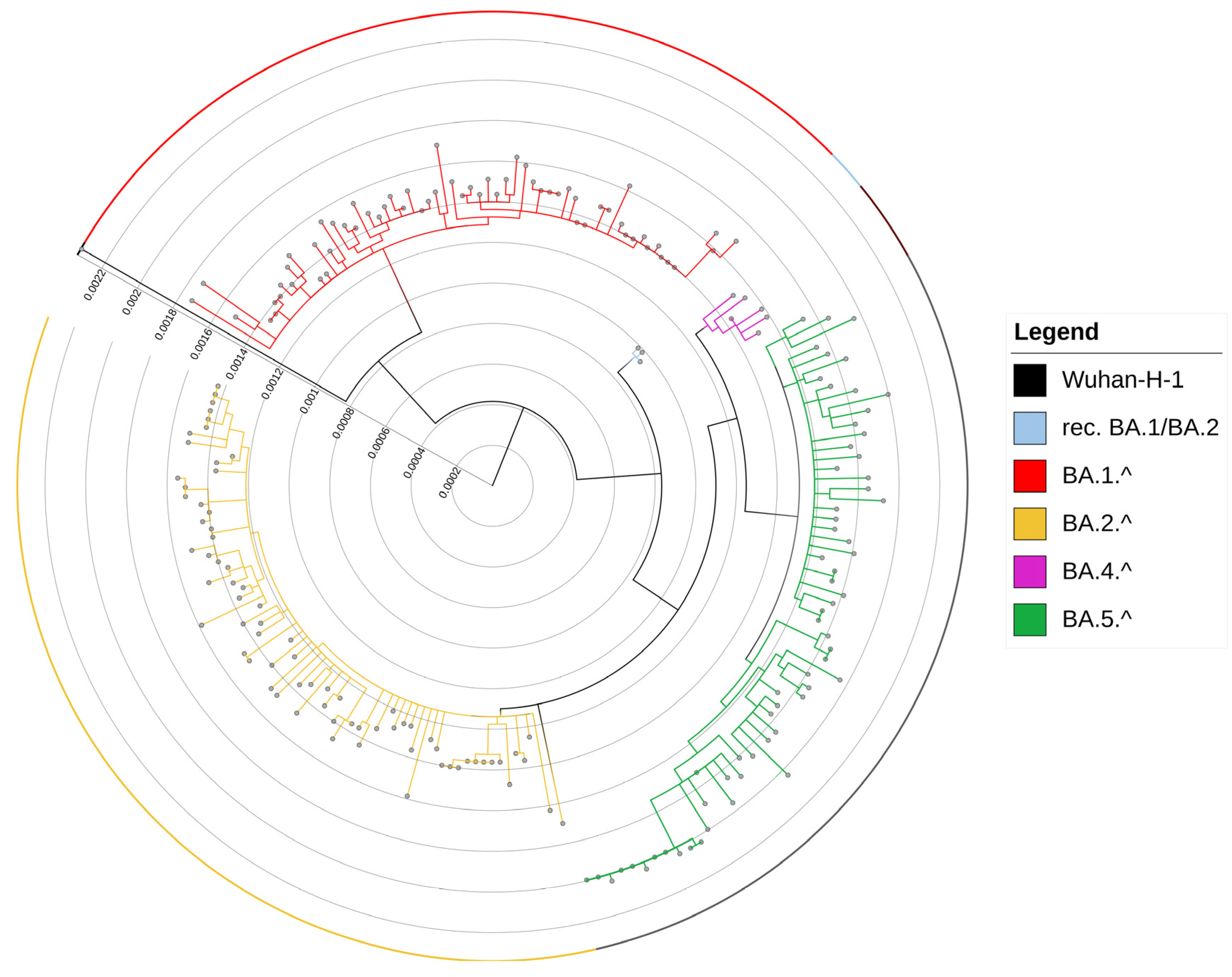
3.1. Evolution of Omicron Sub-Lineages
3.2. Mutations in the S Protein
3.3. Mutations in Structural Proteins
3.4. Mutations in Non-Structural Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coronaviridae Study Group. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e414. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O'Toole, A.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccoli, L.; Park, Y.J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e1021. [Google Scholar] [CrossRef]
- Hirabara, S.M.; Serdan, T.D.A.; Gorjao, R.; Masi, L.N.; Pithon-Curi, T.C.; Covas, D.T.; Curi, R.; Durigon, E.L. SARS-COV-2 Variants: Differences and Potential of Immune Evasion. Front. Cell Infect. Microbiol. 2021, 11, 781429. [Google Scholar] [CrossRef] [PubMed]
- Helmy, Y.A.; Fawzy, M.; Elaswad, A.; Sobieh, A.; Kenney, S.P.; Shehata, A.A. The COVID-19 Pandemic: A Comprehensive Review of Taxonomy, Genetics, Epidemiology, Diagnosis, Treatment, and Control. J. Clin. Med. 2020, 9, 1225. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.; Barrera-Vilarmau, S.; Teixeira, J.M.C.; Sorokina, M.; Seckel, E.; Kastritis, P.L.; Levitt, M. Insights on cross-species transmission of SARS-CoV-2 from structural modeling. PLoS Comput. Biol. 2020, 16, e1008449. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.Q.; Huynh, T. Insights into SARS-CoV-2's Mutations for Evading Human Antibodies: Sacrifice and Survival. J. Med. Chem. 2022, 65, 2820–2826. [Google Scholar] [CrossRef]
- Noureddine, F.Y.; Chakkour, M.; El Roz, A.; Reda, J.; Al Sahily, R.; Assi, A.; Joma, M.; Salami, H.; Hashem, S.J.; Harb, B.; et al. The Emergence of SARS-CoV-2 Variant(s) and Its Impact on the Prevalence of COVID-19 Cases in the Nabatieh Region, Lebanon. Med. Sci. 2021, 9, 40. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.; Peacock, T.P.; Harvey, W.T.; Hughes, J.; Wright, D.W.; Consortium, C.-G.U.; Willett, B.J.; Thomson, E.; Gupta, R.K.; Peacock, S.J.; et al. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat. Rev. Microbiol. 2023, 21, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Banho, C.A.; Sacchetto, L.; Campos, G.R.F.; Bittar, C.; Possebon, F.S.; Ullmann, L.S.; Marques, B.C.; da Silva, G.C.D.; Moraes, M.M.; Parra, M.C.P.; et al. Impact of SARS-CoV-2 Gamma lineage introduction and COVID-19 vaccination on the epidemiological landscape of a Brazilian city. Commun. Med. 2022, 2, 41. [Google Scholar] [CrossRef] [PubMed]
- Escalera, A.; Gonzalez-Reiche, A.S.; Aslam, S.; Mena, I.; Laporte, M.; Pearl, R.L.; Fossati, A.; Rathnasinghe, R.; Alshammary, H.; van de Guchte, A.; et al. Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell Host Microbe 2022, 30, 373–387.e377. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.; Archer, B.; Laurenson-Schafer, H.; Jinnai, Y.; Konings, F.; Batra, N.; Pavlin, B.; Vandemaele, K.; Van Kerkhove, M.D.; Jombart, T.; et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Eurosurveillance 2021, 26, 2100509. [Google Scholar] [CrossRef]
- Fisman, D.N.; Tuite, A.R. Evaluation of the relative virulence of novel SARS-CoV-2 variants: A retrospective cohort study in Ontario, Canada. Can. Med. Assoc. J. 2021, 193, E1619–E1625. [Google Scholar] [CrossRef]
- Available online: https://www.who.int/ (accessed on 30 September 2022).
- Gao, S.J.; Guo, H.T.; Luo, G.X. Omicron variant (B.1.1.529) of SARS-CoV-2, a global urgent public health alert! J. Med. Virol. 2022, 94, 1255–1256. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Xia, S.; Wang, L.J.; Zhu, Y.; Lu, L.; Jiang, S.B. Origin, virological features, immune evasion and intervention of SARS-CoV-2 Omicron sublineages. Signal Transduct. Target. Ther. 2022, 7, 241. [Google Scholar] [CrossRef] [PubMed]
- Stefanelli, P.; Trentini, F.; Petrone, D.; Mammone, A.; Ambrosio, L.; Manica, M.; Guzzetta, G.; d’Andrea, V.; Marziano, V.; Zardini, A.; et al. Tracking the progressive spread of the SARS-CoV-2 Omicron variant in Italy, December 2021—January 2022. Eurosurveillance 2022, 27, 2200125. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.L.; Yisimayi, A.; Jian, F.C.; Song, W.L.; Xiao, T.H.; Wang, L.; Du, S.; Wang, J.; Li, Q.Q.; Chen, X.S.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593. [Google Scholar] [CrossRef] [PubMed]
- Qu, P.K.; Faraone, J.; Evans, J.P.; Zou, X.; Zheng, Y.M.; Carlin, C.; Bednash, J.S.; Lozanski, G.; Mallampalli, R.K.; Saif, L.J.; et al. Neutralization of the SARS-CoV-2 Omicron BA.4/5 and BA.2.12.1 Subvariants. N. Engl. J. Med. 2022, 386, 2526–2528. [Google Scholar] [CrossRef]
- Yuan, S.F.; Ye, Z.W.; Liang, R.H.; Tang, K.M.; Zhang, A.J.; Lu, G.; Ong, C.P.; Poon, V.K.M.; Chan, C.C.S.; Mok, B.W.Y.; et al. Pathogenicity, transmissibility, and fitness of SARS-CoV-2 Omicron in Syrian hamsters. Science 2022, 377, 428–432. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Where did ‘weird’ Omicron come from? Science 2021, 374, 1179. [Google Scholar] [CrossRef]
- Kandeel, M.; Mohamed, M.E.M.; Abd El-Lateef, H.M.; Venugopala, K.N.; El-Beltagi, H.S. Omicron variant genome evolution and phylogenetics. J. Med. Virol. 2022, 94, 1627–1632. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct. Target. Ther. 2022, 7, 138. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ema.europa.eu/ (accessed on 30 September 2022).
- Available online: https://cov-lineages.org/ (accessed on 30 September 2022).
- Lemoine, F.; Correia, D.; Lefort, V.; Doppelt-Azeroual, O.; Mareuil, F.; Cohen-Boulakia, S.; Gascuel, O. NGPhylogeny.fr: New generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019, 47, W260–W265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://itol.embl.de (accessed on 30 September 2022).
- Available online: https://www.epicentro.iss.it (accessed on 30 September 2022).
- De Marco, C.; Veneziano, C.; Massacci, A.; Pallocca, M.; Marascio, N.; Quirino, A.; Barreca, G.S.; Giancotti, A.; Gallo, L.; Lamberti, A.G.; et al. Dynamics of Viral Infection and Evolution of SARS-CoV-2 Variants in the Calabria Area of Southern Italy. Front. Microbiol. 2022, 13, 934993. [Google Scholar] [CrossRef]
- Available online: www.ncbi.nlm.nih.gov (accessed on 30 September 2022).
- Available online: https://outbreak.info/ (accessed on 30 September 2022).
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.C.; Korber, B.; Gnanakaran, S. The SARS-CoV-2 Spike variant D614G favors an open conformational state. Sci. Adv. 2021, 7, eabf3671. [Google Scholar] [CrossRef]
- McCallum, M.; De Marco, A.; Lempp, F.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.M.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332. [Google Scholar] [CrossRef]
- Wang, Z.J.; Muecksch, F.; Cho, A.; Gaebler, C.; Hoffmann, H.H.; Ramos, V.; Zong, S.; Cipolla, M.; Johnson, B.; Schmidt, F.; et al. Analysis of memory B cells identifies conserved neutralizing epitopes on the N-terminal domain of variant SARS-CoV-2 spike proteins. Immunity 2022, 55, 998. [Google Scholar] [CrossRef]
- Wang, Q.; Li, Z.; Ho, J.; Guo, Y.; Yeh, A.Y.; Mohri, H.; Liu, M.; Wang, M.; Yu, J.; Shah, J.G.; et al. Resistance of SARS-CoV-2 omicron subvariant BA.4.6 to antibody neutralisation. Lancet Infect Dis 2022, 22, 1666–1668. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef] [PubMed]
- Gorgun, D.; Lihan, M.; Kapoor, K.; Tajkhorshid, E. Binding mode of SARS-CoV-2 fusion peptide to human cellular membrane. Biophys. J. 2021, 120, 2914–2926. [Google Scholar] [CrossRef] [PubMed]
- Satarker, S.; Nampoothiri, M. Structural Proteins in Severe Acute Respiratory Syndrome Coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Karim, F.; Ganga, Y.; Bernstein, M.; Jule, Z.; Reedoy, K.; Cele, S.; Lustig, G.; Amoako, D.; Wolter, N.; et al. Omicron BA.4/BA.5 escape neutralizing immunity elicited by BA.1 infection. Nat. Commun. 2022, 13, 4686. [Google Scholar] [CrossRef]
- Jian, M.J.; Chung, H.Y.; Chang, C.K.; Lin, J.C.; Yeh, K.M.; Chen, C.W.; Lin, D.Y.; Chang, F.Y.; Hung, K.S.; Perng, C.L.; et al. SARS-CoV-2 variants with T135I nucleocapsid mutations may affect antigen test performance. Int. J. Infect. Dis. 2022, 114, 112–114. [Google Scholar] [CrossRef]
- Mourier, T.; Shuaib, M.; Hala, S.; Mfarrej, S.; Alofi, F.; Naeem, R.; Alsomali, A.; Jorgensen, D.; Subudhi, A.K.; Ben Rached, F.; et al. SARS-CoV-2 genomes from Saudi Arabia implicate nucleocapsid mutations in host response and increased viral load. Nat. Commun. 2022, 13, 601. [Google Scholar] [CrossRef]
- Russo, A.; Serapide, F.; Quirino, A.; Tarsitano, M.G.; Marascio, N.; Serraino, R.; Rotundo, S.; Matera, G.; Trecarichi, E.M.; Torti, C. Microbiological and Clinical Findings of SARS-CoV-2 Infection after 2 Years of Pandemic: From Lung to Gut Microbiota. Diagnostics 2022, 12, 2143. [Google Scholar] [CrossRef]
- Shannon, A.; Le, N.T.T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemot, J.C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 Exonuclease active-sites. Antivir. Res. 2020, 178, 104793. [Google Scholar] [CrossRef]
- Badua, C.L.D.C.; Baldo, K.A.T.; Medina, P.M.B. Genomic and proteomic mutation landscapes of SARS-CoV-2. J. Med. Virol. 2021, 93, 1702–1721. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.M.; Huang, Y.C.; Liu, F.J.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.Q.; Ming, Z.H.; Zhang, L.Q.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Balaji, S.; Lomakin, I.B.; Xiong, Y. Coronavirus Nsp1: Immune Response Suppression and Protein Expression Inhibition. Front. Microbiol. 2021, 12, 752214. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Makino, S. Mechanisms of Coronavirus Nsp1-Mediated Control of Host and Viral Gene Expression. Cells 2021, 10, 300. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.W.; Tang, C.; Wei, H.C.; Du, B.W.; Chen, C.; Wang, M.J.; Zhou, Y.Z.; Yu, M.X.; Cheng, L.; Kuivanen, S.; et al. Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response. Cell Host Microbe 2021, 29, 489. [Google Scholar] [CrossRef] [PubMed]
- Sk, S.H.A.; Pabitra, P.C.B.; Guy, W.D.I.C.; Aljabali, A.A.A.; Uhal, B.D.; Lundstrom, K.; Rezaei, N.; Pizzol, D.; Adadi, P.; Lal, A.; et al. The importance of accessory protein variants in the pathogenicity of SARS-CoV-2. Arch. Biochem. Biophys. 2022, 717, 109124. [Google Scholar] [CrossRef]
- Giri, R.; Bhardwaj, T.; Shegane, M.; Gehi, B.R.; Kumar, P.; Gadhave, K.; Oldfield, C.J.; Uversky, V.N. Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronaviruses. Cell. Mol. Life Sci. 2021, 78, 1655–1688. [Google Scholar] [CrossRef]
- Kern, D.M.; Sorum, B.; Mali, S.S.; Hoel, C.M.; Sridharan, S.; Remis, J.P.; Toso, D.B.; Kotecha, A.; Bautista, D.M.; Brohawn, S.G. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 702. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Borsetti, A.; Ciccozzi, M.; Pascarella, S. SARS-Cov-2 ORF3a: Mutability and function. Int. J. Biol. Macromol. 2021, 170, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Flower, T.G.; Buffalo, C.Z.; Hooy, R.M.; Allaire, M.; Ren, X.F.; Hurley, J.H. Structure of SARS-CoV-2 ORF8, a rapidly evolving immune evasion protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2021785118. [Google Scholar] [CrossRef]
- Hassan, S.S.; Aljabali, A.A.A.; Panda, P.K.; Ghosh, S.; Attrish, D.; Choudhury, P.P.; Seyran, M.; Pizzol, D.; Adadi, P.; Abd El-Aziz, T.M.; et al. A unique view of SARS-CoV-2 through the lens of ORF8 protein. Comput. Biol. Med. 2021, 133, 104380. [Google Scholar] [CrossRef]
- Pereira, F. Evolutionary dynamics of the SARS-CoV-2 ORF8 accessory gene. Infect. Genet. Evol. 2020, 85, 104525. [Google Scholar] [CrossRef]
- Gong, F.; Wei, H.X.; Li, Q.; Liu, L.; Li, B. Evaluation and Comparison of Serological Methods for COVID-19 Diagnosis. Front. Mol. Biosci. 2021, 8, 682405. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.F.; Anderson, D.E.; Young, B.E.; Linster, M.; Zhu, F.; Jayakumar, J.; Zhuang, Y.; Kalimuddin, S.; Low, J.G.H.; Tan, C.W.; et al. Discovery and Genomic Characterization of a 382-Nucleotide Deletion in ORF7b and ORF8 during the Early Evolution of SARS-CoV-2. mBio 2020, 11, e01610-20. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Percentage (%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Recombinant BA.1/BA.2 | BA.1 | BA.1.1 | BA.2 | BA.2.9 | BA.2.10 | BA.2.12.1 | BA.4 | BA.5 | BE.1 | Other | ||
| January | Calabria | 0 | 78 | 22 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Italy | 0 | 66 | 33 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| February | Calabria | 0 | 49 | 51 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Italy | 0 | 42 | 44 | 11 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | |
| March | Calabria | 0 | 22 | 49 | 29 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Italy | 0 | 13 | 22 | 53 | 11 | 1 | 0 | 0 | 0 | 0 | 0 | |
| April | Calabria | 13 | 0 | 13 | 66 | 6 | 3 | 0 | 0 | 0 | 0 | 0 |
| Italy | 0 | 2 | 7 | 74 | 16 | 1 | 0 | 0 | 0 | 0 | 0 | |
| May | Calabria | 0 | 0 | 0 | 45 | 55 | 0 | 0 | 0 | 0 | 0 | 0 |
| Italy | 0 | 1 | 1 | 78 | 14 | 1 | 1 | 2 | 2 | 0 | 0 | |
| June | Calabria | 0 | 0 | 0 | 59 | 29 | 0 | 0 | 6 | 6 | 0 | 0 |
| Italy | 0 | 0 | 0 | 34 | 5 | 0 | 4 | 13 | 37 | 5 | 2 | |
| July | Calabria | 0 | 0 | 0 | 14 | 2 | 0 | 4 | 9 | 71 | 0 | 0 |
| Italy | 0 | 0 | 0 | 5 | 1 | 0 | 1 | 11 | 71 | 9 | 2 | |
| August | Calabria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 13 | 88 | 0 | 0 |
| Italy | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 7 | 79 | 7 | 5 | |
| September | Calabria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 94 | 6 | 0 |
| Italy | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 4 | 78 | 7 | 9 | |
| Non-Canonical Mutations | Isolates with Non-Canonical Mutations N/368 | Isolates with Non-Canonical Mutations % | Omicron Lineages |
|---|---|---|---|
| L5F | 9 | 2.5 | BA.2.^; BA.5.^ |
| V6I | 1 | 0.3 | BA.2.^ |
| P9T | 1 | 0.3 | BA.1.^ |
| S12F | 1 | 0.3 | BA.5.^ |
| T22I | 1 | 0.3 | BA.1.^ |
| M153I | 1 | 0.3 | BA.5.^ |
| D178N | 1 | 0.3 | BA.4.^ |
| G181V | 1 | 0.3 | BA.5.^ |
| G181A | 1 | 0.3 | BA.5.^ |
| P209L | 1 | 0.3 | BA.5.^ |
| I624M | 1 | 0.3 | BA.2.^ |
| N658S | 3 | 0.8 | BA.4.^ |
| A701V | 10 | 2.7 | BA.1.^ |
| A713S | 2 | 0.5 | BA.5.^ |
| P862S | 1 | 0.3 | BA.5.^ |
| E1144V | 1 | 0.3 | BA.5.^ |
| Subdomain of S Protein | Size of Subdomain | Number of Non-Canonical Mutations Observed in Subdomain | Mutation Density (%) |
|---|---|---|---|
| NTD | 291 aa | 10 (1 in BA.1.^; 2 in BA.2.^; 1 in BA.4.^; 7 in BA.5.^) | 3.4 |
| RBD | 196 aa | 0 | 0.0 |
| CT1–CT2 | 157 aa | 2 (1 in BA.2.^; 1 in BA.4.^) | 1.3 |
| S1/S2 cleavage | 129 aa | 2 (1 in BA.1.^; 1 in BA.5.^) | 1.6 |
| FP | 95 aa | 1 in BA.5.^ | 1.1 |
| HR1 | 65 aa | 0 | 0.0 |
| CD1 | 76 aa | 1 in BA.5.^ | 1.3 |
| Gene | Protein (aa) | Non-Canonical Mutations | Isolates with Non-Canonical Mutations |
|---|---|---|---|
| ORF1ab | nsp1 (1–180) | 141–143 del | 27 (13%) |
| 82–86 del | |||
| 85 del | |||
| V38F, G82C/D, L92V, L107F, V111M, K120N, R124H, N178S | |||
| nsp2 (181–818) | L642F, G392C, S318L, G519S, L624F, G697R, R207C, A566V, A486V, H417Y, S302F, A239V, P361T, P309L, P626L, R545Q, H374Y, A498V, P361S, A690V, T814I, V559A, L204F, A599V, L454F, Q556K, I431M, A801V | 37; (18%) | |
| nsp3 (819–2763) | T1597I, V1211F, S1534I, P1803S, S1612L, T1760I, K1929R, A1473V, L2146F, M1083I, S1188L, H1160Y, A1997V, A1631V, T1496I, G1073V, P1220L, A1809T, T1242I, T1444A, V2116L, Y1465CM, H1545Y, L1450F, D1507N, P2046L, D2037N, A1204T | 31; (15%) | |
| nsp4 | S2797F, P2929L, A2784V, W2769R, T2823I, N2272S, A2828V | 8; (4%) | |
| nsp6 | I3758V, T3750I, P3767S, L3796F, L3829F | 10; (6%) | |
| nsp12 (4393–5324) | I4498V + E4661N | 9; (4%) | |
| L5141M, H4474Y, G4436V, N4473E, K4483R, I4563M, R4565C, T5131I | |||
| nsp13 (5325–5925) | S5362P | 10; (5%) | |
| S5360P + D5693G | |||
| A5703V, S5360P, S5560C, S5398P, M5557I, P5853L, Y5601H | |||
| nsp14 (5926–6452) | A6532V + V6579F | 13; (6%) | |
| P6128L + G6013S | |||
| H6208Q + T6564I | |||
| P6128L, T5941I, G6621R, A6612V, L6082F, L6614F, V6492I, V6107I, V6474L, R6590L | |||
| nsp15 (6453–6798) | H6789Y | 1; (0.5%) | |
| ORF3a | ORF3a (1–275) | 256–259 del | 15; (7%) |
| L52F + F207L | |||
| V13A, A54V, R68G, L106F, L108F, R122I, S171L, E239D | |||
| M | Membrane (1–222) | S4F, L34F, T7I, A85V, H125R | 6; (3%) |
| ORF6 | ORF6 (1–61) | S41C | 1; (0.5%) |
| ORF7a | ORF7a (1–121) | L9M, F101fs | 2; (1%) |
| ORF8 | ORF8 (1–121) | Q27 *, A65D, S67F, E64G | 12; (6%) |
| N | Nucleocapsid (1–419) | D3V, T49I, H59R, D103N, P162H, N181L, S194L, A220T, G275C, T362I, P365S, T366I, T379I, A414S | 17; (8%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veneziano, C.; Marascio, N.; De Marco, C.; Quaresima, B.; Biamonte, F.; Trecarichi, E.M.; Santamaria, G.; Quirino, A.; Torella, D.; Quattrone, A.; et al. The Spread of SARS-CoV-2 Omicron Variant in CALABRIA: A Spatio-Temporal Report of Viral Genome Evolution. Viruses 2023, 15, 408. https://doi.org/10.3390/v15020408
Veneziano C, Marascio N, De Marco C, Quaresima B, Biamonte F, Trecarichi EM, Santamaria G, Quirino A, Torella D, Quattrone A, et al. The Spread of SARS-CoV-2 Omicron Variant in CALABRIA: A Spatio-Temporal Report of Viral Genome Evolution. Viruses. 2023; 15(2):408. https://doi.org/10.3390/v15020408
Chicago/Turabian StyleVeneziano, Claudia, Nadia Marascio, Carmela De Marco, Barbara Quaresima, Flavia Biamonte, Enrico Maria Trecarichi, Gianluca Santamaria, Angela Quirino, Daniele Torella, Aldo Quattrone, and et al. 2023. "The Spread of SARS-CoV-2 Omicron Variant in CALABRIA: A Spatio-Temporal Report of Viral Genome Evolution" Viruses 15, no. 2: 408. https://doi.org/10.3390/v15020408
APA StyleVeneziano, C., Marascio, N., De Marco, C., Quaresima, B., Biamonte, F., Trecarichi, E. M., Santamaria, G., Quirino, A., Torella, D., Quattrone, A., Matera, G., Torti, C., De Filippo, C., Costanzo, F. S., & Viglietto, G. (2023). The Spread of SARS-CoV-2 Omicron Variant in CALABRIA: A Spatio-Temporal Report of Viral Genome Evolution. Viruses, 15(2), 408. https://doi.org/10.3390/v15020408