
Viral Hemorrhagic Septicemia Virus Activates Integrated Stress Response Pathway and Induces Stress Granules to Regulate Virus Replication

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. VHSV Infection
2.3. VHSV Amplification and Titering
2.4. Chemicals, Plasmids and Antibodies
2.5. Cell Treatments
2.6. Immunoblotting
2.7. Immunofluorescence Microscopy
2.8. Real-Time Quantitative PCR
2.9. CRISPR Knockdown Cell Line
2.10. Transfection
2.11. Luciferase Assay
2.12. Virus Yield and IFN Bioassay
2.13. Statistics
3. Results
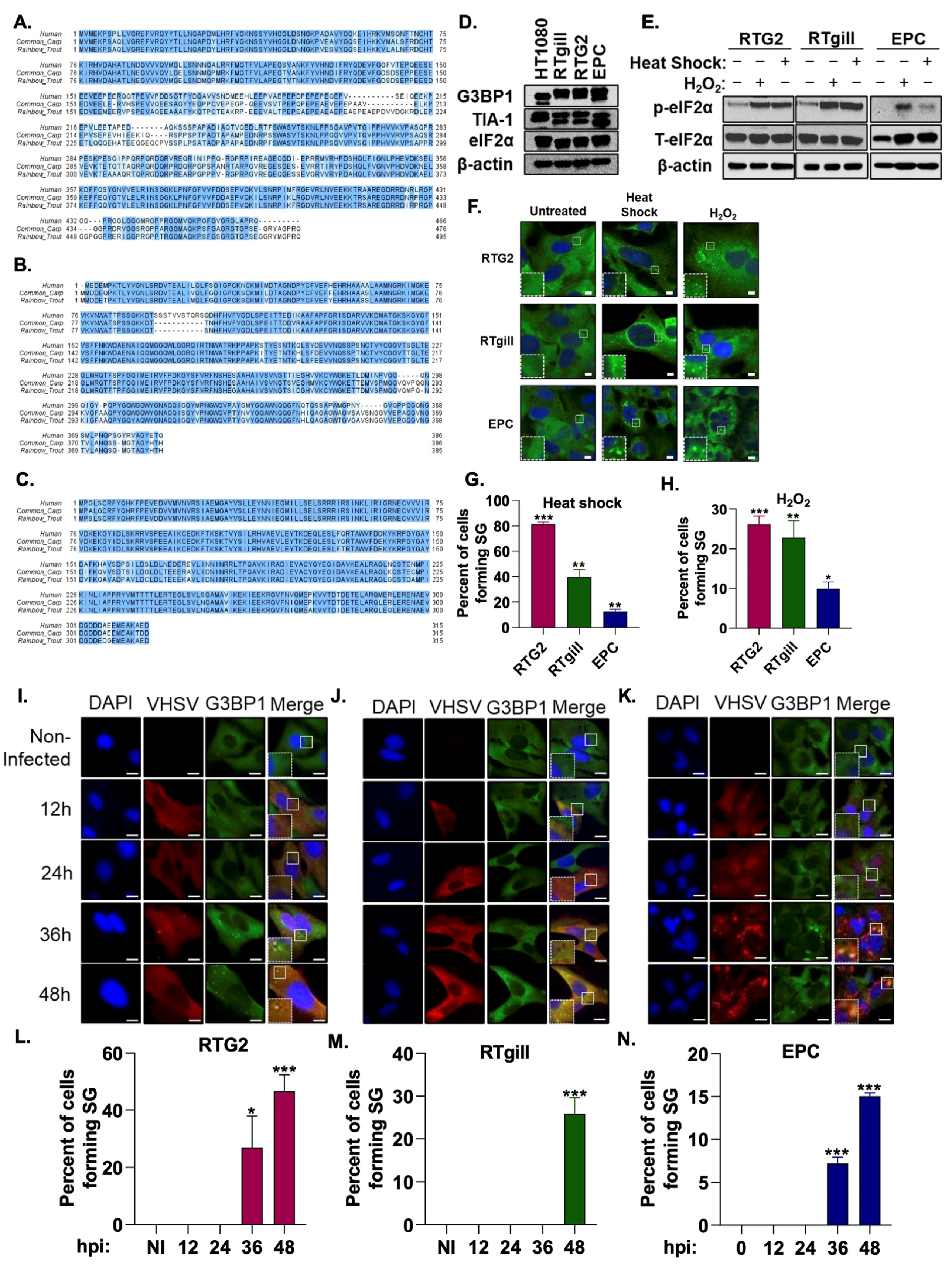
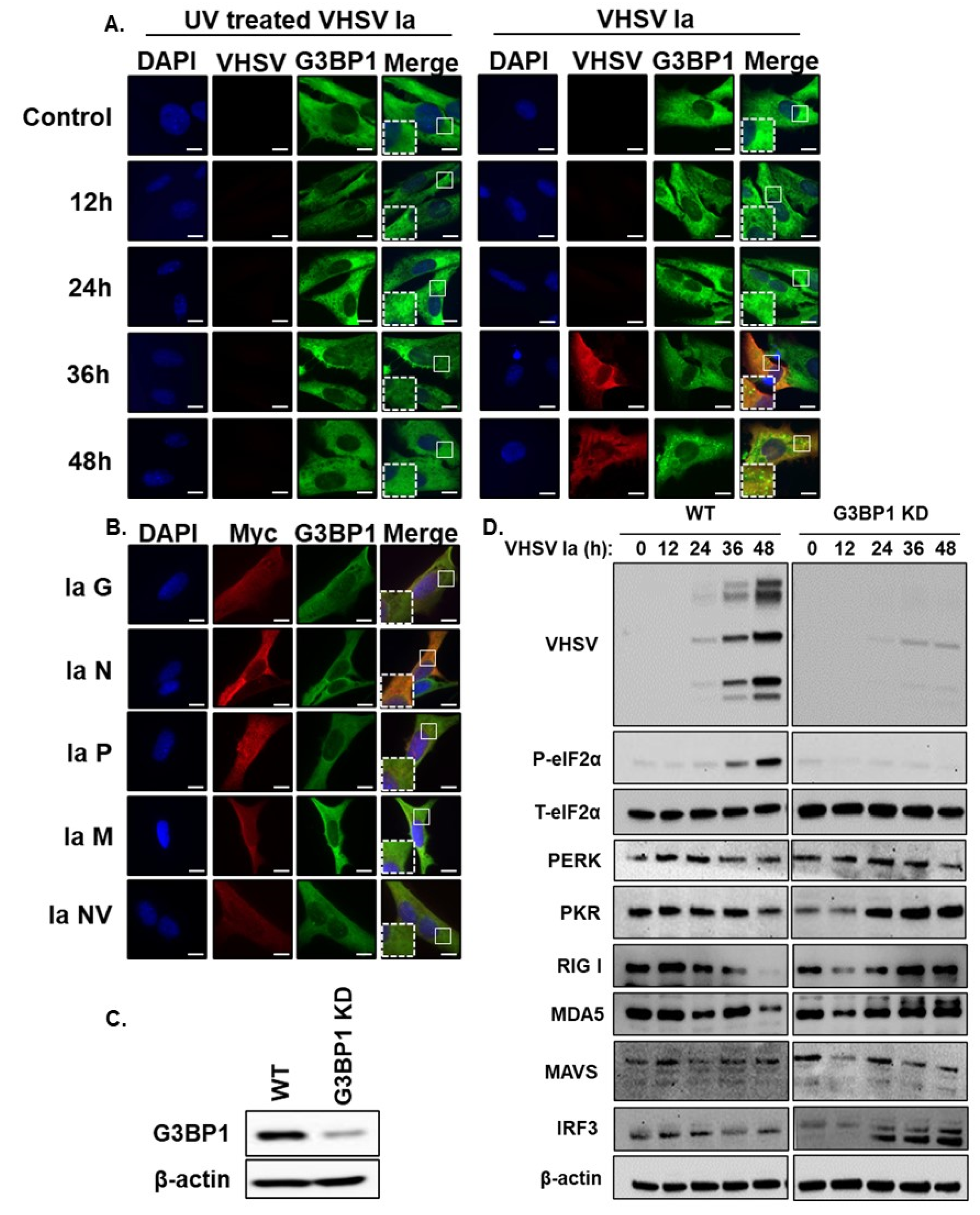
3.1. VHSV Infection Induces Stress Granule Formation
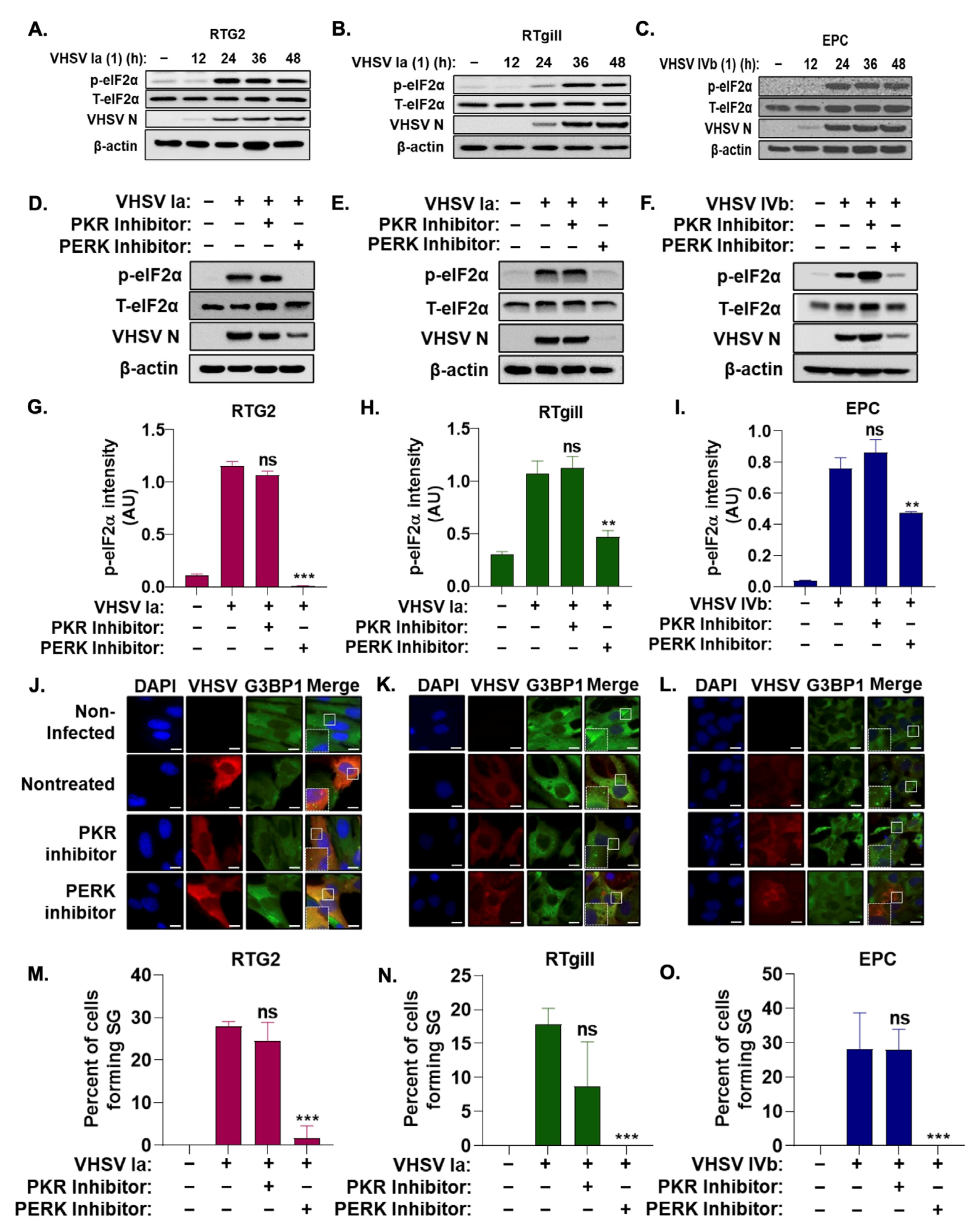
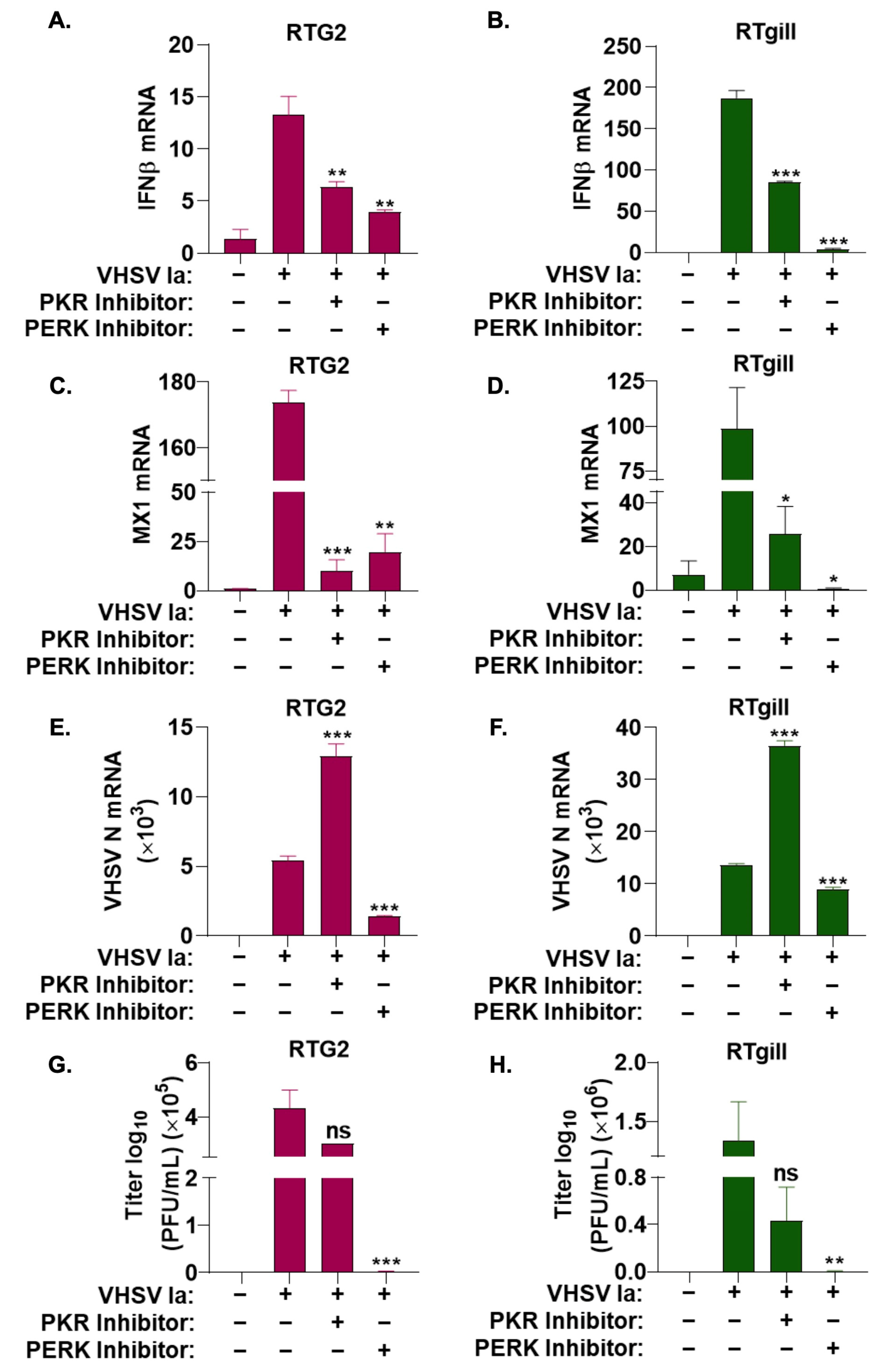
3.2. Stress Granule Formation Requires PERK Activation
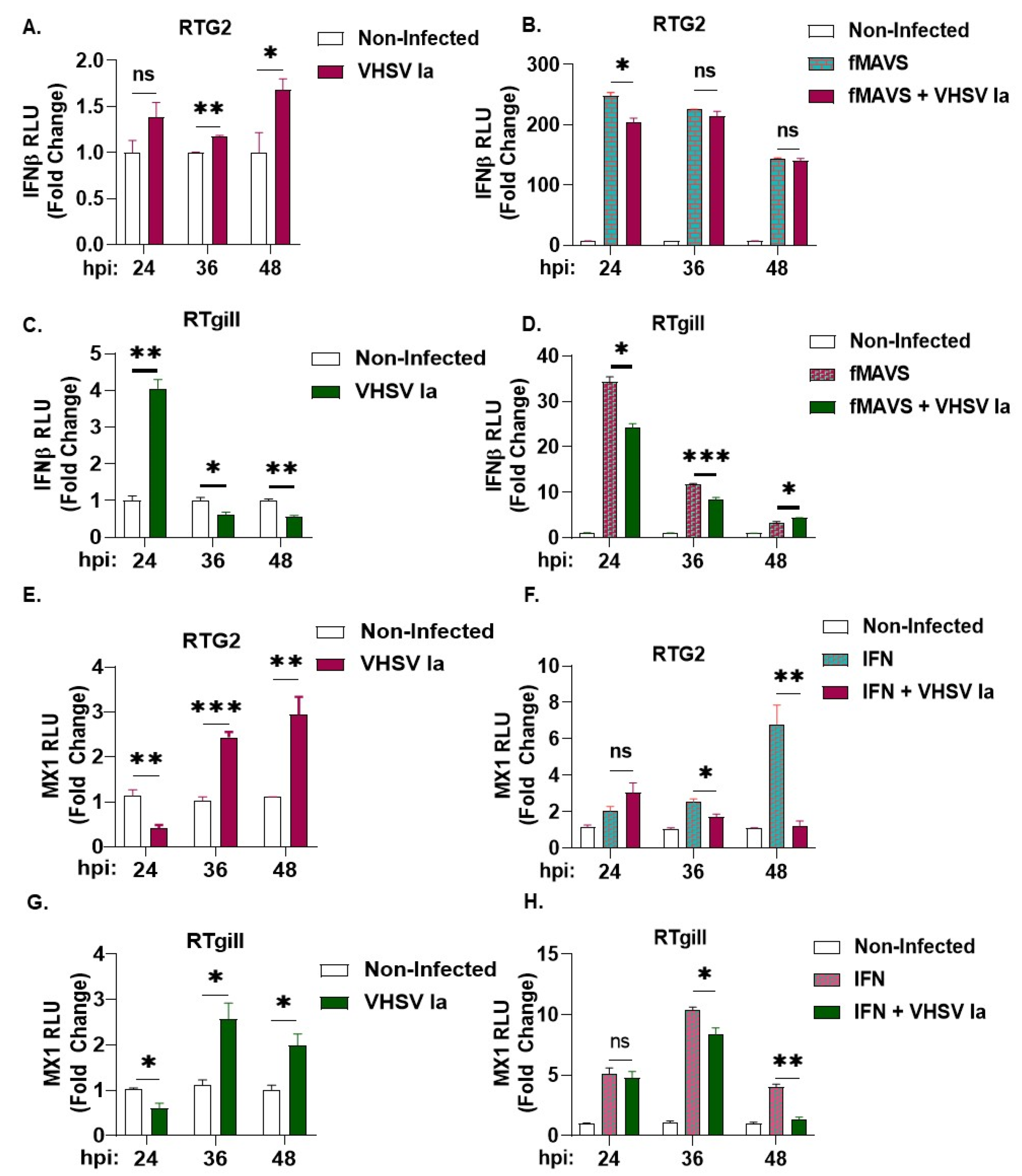
3.3. Role of Integrated Stress Response on IFN Signaling during VHSV Infection
3.4. Viral Replication Is Required for SG Formation
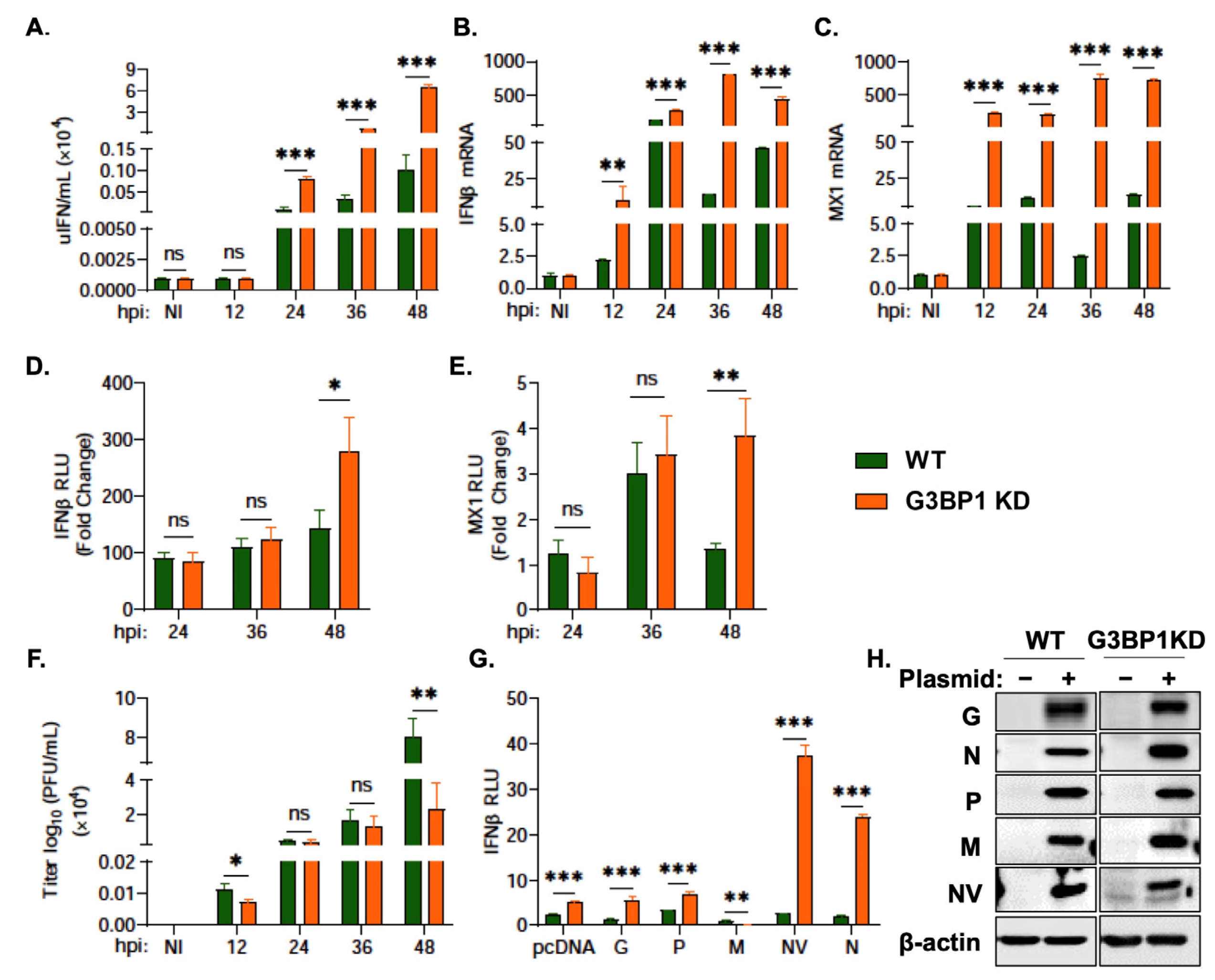
3.5. G3BP1 Regulates IFN Signaling during VHSV Ia Infection
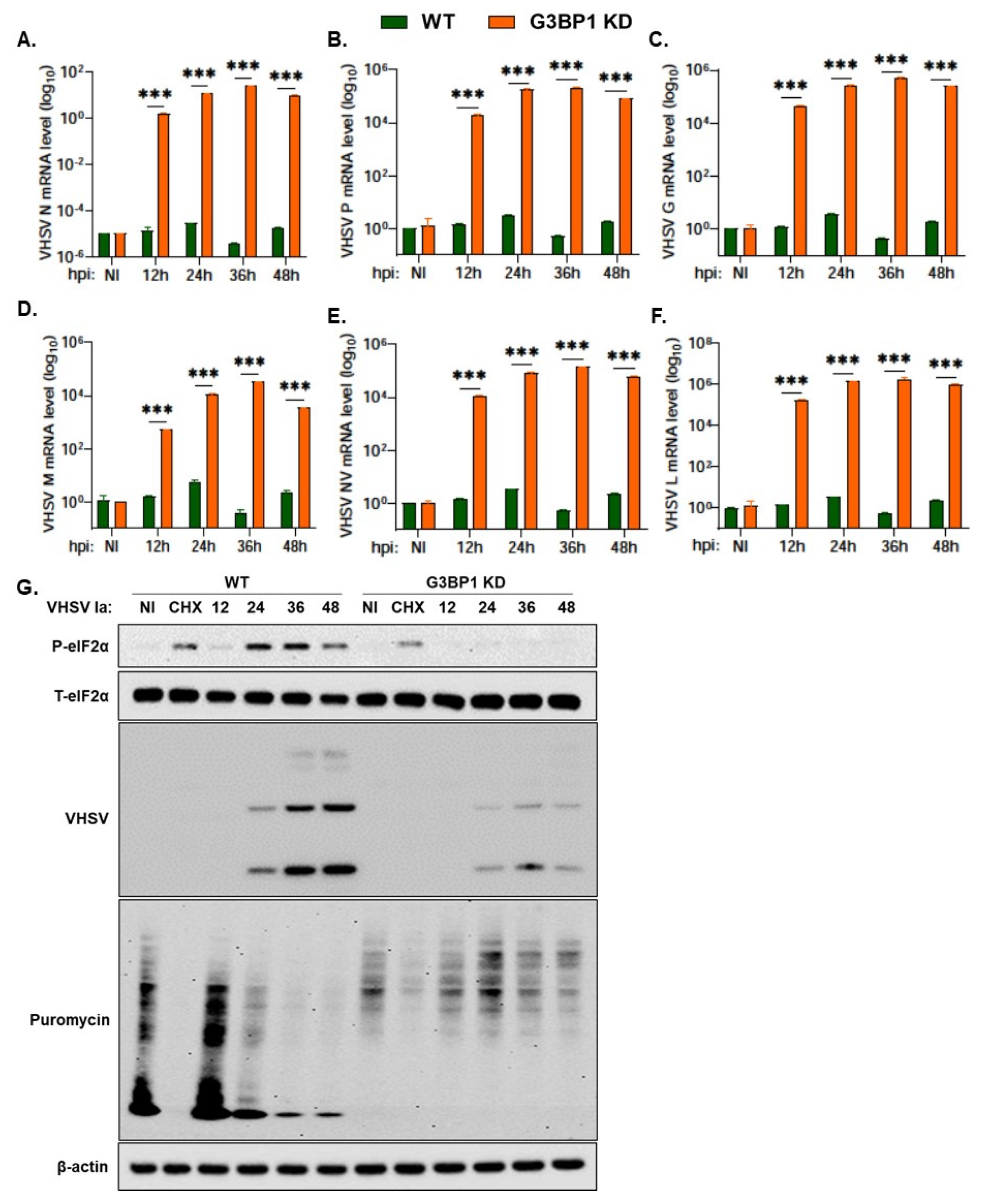
3.6. G3BP1 Is Required for Viral Protein Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational regulation of gene expression during conditions of cell stress. Mol. Cell 2010, 40, 228–237. [Google Scholar] [CrossRef]
- Liu, B.; Qian, S.B. Translational reprogramming in cellular stress response. Wiley Interdiscip. Rev. RNA 2014, 5, 301–315. [Google Scholar] [CrossRef]
- Brostrom, C.O.; Prostko, C.R.; Kaufman, R.J.; Brostrom, M.A. Inhibition of translational initiation by activators of the glucose-regulated stress protein and heat shock protein stress response systems. Role of the interferon-inducible double-stranded RNA-activated eukaryotic initiation factor 2alpha kinase. J. Biol. Chem. 1996, 271, 24995–25002. [Google Scholar] [CrossRef]
- Stern-Ginossar, N.; Thompson, S.R.; Mathews, M.B.; Mohr, I. Translational Control in Virus-Infected Cells. Cold Spring Harb. Perspect. Biol. 2019, 11, a033001. [Google Scholar] [CrossRef]
- Walsh, D.; Mathews, M.B.; Mohr, I. Tinkering with translation: Protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 2013, 5, a012351. [Google Scholar] [CrossRef]
- Piccirillo, C.A.; Bjur, E.; Topisirovic, I.; Sonenberg, N.; Larsson, O. Translational control of immune responses: From transcripts to translatomes. Nat. Immunol. 2014, 15, 503–511. [Google Scholar] [CrossRef]
- McCormick, C.; Khaperskyy, D.A. Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 2017, 17, 647–660. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2alpha kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Perkins, D.J.; Barber, G.N. Defects in translational regulation mediated by the alpha subunit of eukaryotic initiation factor 2 inhibit antiviral activity and facilitate the malignant transformation of human fibroblasts. Mol. Cell. Biol. 2004, 24, 2025–2040. [Google Scholar] [CrossRef]
- Balachandran, S.; Barber, G.N. PKR in innate immunity, cancer, and viral oncolysis. Methods Mol. Biol. 2007, 383, 277–301. [Google Scholar]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahboubi, H.; Stochaj, U. Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Khong, A.; Matheny, T.; Jain, S.; Mitchell, S.F.; Wheeler, J.R.; Parker, R. The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules. Mol. Cell 2017, 68, 808–820 e805. [Google Scholar] [CrossRef] [PubMed]
- Namkoong, S.; Ho, A.; Woo, Y.M.; Kwak, H.; Lee, J.H. Systematic Characterization of Stress-Induced RNA Granulation. Mol. Cell 2018, 70, 175–187 e178. [Google Scholar] [CrossRef] [PubMed]
- Van Treeck, B.; Protter, D.S.W.; Matheny, T.; Khong, A.; Link, C.D.; Parker, R. RNA self-assembly contributes to stress granule formation and defining the stress granule transcriptome. Proc. Natl. Acad. Sci. USA 2018, 115, 2734–2739. [Google Scholar] [CrossRef]
- Youn, J.Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.R.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.M.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532 e511. [Google Scholar] [CrossRef]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef]
- Tourriere, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003, 160, 823–831. [Google Scholar] [CrossRef]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct stages in stress granule assembly and disassembly. Elife 2016, 5, 18413. [Google Scholar] [CrossRef]
- Aulas, A.; Fay, M.M.; Lyons, S.M.; Achorn, C.A.; Kedersha, N.; Anderson, P.; Ivanov, P. Stress-specific differences in assembly and composition of stress granules and related foci. J. Cell Sci. 2017, 130, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef]
- Manivannan, P.; Siddiqui, M.A.; Malathi, K. RNase L Amplifies Interferon Signaling by Inducing Protein Kinase R-Mediated Antiviral Stress Granules. J. Virol. 2020, 94, e00205-20. [Google Scholar] [CrossRef]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PloS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Rozelle, D.K.; Filone, C.M.; Kedersha, N.; Connor, J.H. Activation of stress response pathways promotes formation of antiviral granules and restricts virus replication. Mol. Cell. Biol. 2014, 34, 2003–2016. [Google Scholar] [CrossRef]
- Yoneyama, M.; Jogi, M.; Onomoto, K. Regulation of antiviral innate immune signaling by stress-induced RNA granules. J. Biochem. 2016, 159, 279–286. [Google Scholar] [CrossRef]
- Yoo, J.S.; Takahasi, K.; Ng, C.S.; Ouda, R.; Onomoto, K.; Yoneyama, M.; Lai, J.C.; Lattmann, S.; Nagamine, Y.; Matsui, T.; et al. DHX36 enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog. 2014, 10, e1004012. [Google Scholar] [CrossRef]
- Thulasi Raman, S.N.; Liu, G.; Pyo, H.M.; Cui, Y.C.; Xu, F.; Ayalew, L.E.; Tikoo, S.K.; Zhou, Y. DDX3 Interacts with Influenza A Virus NS1 and NP Proteins and Exerts Antiviral Function through Regulation of Stress Granule Formation. J. Virol. 2016, 90, 3661–3675. [Google Scholar] [CrossRef]
- White, J.P.; Cardenas, A.M.; Marissen, W.E.; Lloyd, R.E. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2007, 2, 295–305. [Google Scholar] [CrossRef]
- Panas, M.D.; Varjak, M.; Lulla, A.; Eng, K.E.; Merits, A.; Karlsson Hedestam, G.B.; McInerney, G.M. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol. Biol. Cell 2012, 23, 4701–4712. [Google Scholar] [CrossRef]
- Yi, Z.; Pan, T.; Wu, X.; Song, W.; Wang, S.; Xu, Y.; Rice, C.M.; Macdonald, M.R.; Yuan, Z. Hepatitis C virus co-opts Ras-GTPase-activating protein-binding protein 1 for its genome replication. J. Virol. 2011, 85, 6996–7004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garaigorta, U.; Heim, M.H.; Boyd, B.; Wieland, S.; Chisari, F.V. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J. Virol. 2012, 86, 11043–11056. [Google Scholar] [CrossRef]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef] [PubMed]
- Brocard, M.; Iadevaia, V.; Klein, P.; Hall, B.; Lewis, G.; Lu, J.; Burke, J.; Willcocks, M.M.; Parker, R.; Goodfellow, I.G.; et al. Norovirus infection results in eIF2alpha independent host translation shut-off and remodels the G3BP1 interactome evading stress granule formation. PLoS Pathog. 2020, 16, e1008250. [Google Scholar] [CrossRef] [PubMed]
- Hosmillo, M.; Lu, J.; McAllaster, M.R.; Eaglesham, J.B.; Wang, X.; Emmott, E.; Domingues, P.; Chaudhry, Y.; Fitzmaurice, T.J.; Tung, M.K.; et al. Noroviruses subvert the core stress granule component G3BP1 to promote viral VPg-dependent translation. Elife 2019, 8, 46681. [Google Scholar] [CrossRef]
- Skall, H.F.; Olesen, N.J.; Mellergaard, S. Viral haemorrhagic septicaemia virus in marine fish and its implications for fish farming—A review. J. Fish Dis. 2005, 28, 509–529. [Google Scholar] [CrossRef]
- Schutze, H.; Enzmann, P.J.; Mundt, E.; Mettenleiter, T.C. Identification of the non-virion (NV) protein of fish rhabdoviruses viral haemorrhagic septicaemia virus and infectious haematopoietic necrosis virus. J. Gen. Virol. 1996, 77 Pt 6, 1259–1263. [Google Scholar] [CrossRef]
- Schutze, H.; Mundt, E.; Mettenleiter, T.C. Complete genomic sequence of viral hemorrhagic septicemia virus, a fish rhabdovirus. Virus Genes 1999, 19, 59–65. [Google Scholar] [CrossRef]
- Ammayappan, A.; Vakharia, V.N. Molecular characterization of the Great Lakes viral hemorrhagic septicemia virus (VHSV) isolate from USA. Virol. J. 2009, 6, 171. [Google Scholar] [CrossRef]
- Snow, M.; Cunningham, C.O.; Melvin, W.T.; Kurath, G. Analysis of the nucleoprotein gene identifies distinct lineages of viral haemorrhagic septicaemia virus within the European marine environment. Virus Res. 1999, 63, 35–44. [Google Scholar] [CrossRef]
- Einer-Jensen, K.; Ahrens, P.; Forsberg, R.; Lorenzen, N. Evolution of the fish rhabdovirus viral haemorrhagic septicaemia virus. J. Gen. Virol. 2004, 85, 1167–1179. [Google Scholar] [CrossRef]
- Stone, D.M.; Ferguson, H.W.; Tyson, P.A.; Savage, J.; Wood, G.; Dodge, M.J.; Woolford, G.; Dixon, P.F.; Feist, S.W.; Way, K. The first report of viral haemorrhagic septicaemia in farmed rainbow trout, Oncorhynchus mykiss (Walbaum), in the United Kingdom. J. Fish Dis. 2008, 31, 775–784. [Google Scholar] [CrossRef]
- Elsayed, E.; Faisal, M.; Thomas, M.; Whelan, G.; Batts, W.; Winton, J. Isolation of viral haemorrhagic septicaemia virus from muskellunge, Esox masquinongy (Mitchill), in Lake St Clair, Michigan, USA reveals a new sublineage of the North American genotype. J. Fish Dis. 2006, 29, 611–619. [Google Scholar] [CrossRef]
- Lumsden, J.S.; Morrison, B.; Yason, C.; Russell, S.; Young, K.; Yazdanpanah, A.; Huber, P.; Al-Hussinee, L.; Stone, D.; Way, K. Mortality event in freshwater drum Aplodinotus grunniens from Lake Ontario, Canada, associated with viral haemorrhagic septicemia virus, type IV. Dis. Aquat. Org. 2007, 76, 99–111. [Google Scholar] [CrossRef]
- Gagne, N.; Mackinnon, A.M.; Boston, L.; Souter, B.; Cook-Versloot, M.; Griffiths, S.; Olivier, G. Isolation of viral haemorrhagic septicaemia virus from mummichog, stickleback, striped bass and brown trout in eastern Canada. J. Fish Dis. 2007, 30, 213–223. [Google Scholar] [CrossRef]
- Altmann, S.M.; Mellon, M.T.; Distel, D.L.; Kim, C.H. Molecular and functional analysis of an interferon gene from the zebrafish, Danio rerio. J. Virol. 2003, 77, 1992–2002. [Google Scholar] [CrossRef]
- Schultz, U.; Kaspers, B.; Staeheli, P. The interferon system of non-mammalian vertebrates. Dev. Comp. Immunol. 2004, 28, 499–508. [Google Scholar] [CrossRef]
- Zou, J.; Tafalla, C.; Truckle, J.; Secombes, C.J. Identification of a second group of type I IFNs in fish sheds light on IFN evolution in vertebrates. J. Immunol. 2007, 179, 3859–3871. [Google Scholar] [CrossRef]
- Zou, J.; Bird, S.; Secombes, C. Antiviral sensing in teleost fish. Curr. Pharm. Des. 2010, 16, 4185–4193. [Google Scholar] [CrossRef]
- Chang, M.; Collet, B.; Nie, P.; Lester, K.; Campbell, S.; Secombes, C.J.; Zou, J. Expression and functional characterization of the RIG-I-like receptors MDA5 and LGP2 in Rainbow trout (Oncorhynchus mykiss). J. Virol. 2011, 85, 8403–8412. [Google Scholar] [CrossRef]
- Biacchesi, S.; LeBerre, M.; Lamoureux, A.; Louise, Y.; Lauret, E.; Boudinot, P.; Bremont, M. Mitochondrial antiviral signaling protein plays a major role in induction of the fish innate immune response against RNA and DNA viruses. J. Virol. 2009, 83, 7815–7827. [Google Scholar] [CrossRef] [Green Version]
- Verrier, E.R.; Langevin, C.; Benmansour, A.; Boudinot, P. Early antiviral response and virus-induced genes in fish. Dev. Comp. Immunol. 2011, 35, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Weaver, W.; Pore, A.; Gorgoglione, B.; Wildschutte, J.H.; Xiao, P.; Shepherd, B.S.; Spear, A.; Malathi, K.; Stepien, C.A.; et al. Role of Viral Hemorrhagic Septicemia Virus Matrix (M) Protein in Suppressing Host Transcription. J. Virol. 2017, 91, e00279-17. [Google Scholar] [CrossRef] [PubMed]
- Biacchesi, S.; Merour, E.; Chevret, D.; Lamoureux, A.; Bernard, J.; Bremont, M. NV Proteins of Fish Novirhabdovirus Recruit Cellular PPM1Bb Protein Phosphatase and Antagonize RIG-I-Mediated IFN Induction. Sci. Rep. 2017, 7, 44025. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, K.H. Effects of NV gene knock-out recombinant viral hemorrhagic septicemia virus (VHSV) on Mx gene expression in Epithelioma papulosum cyprini (EPC) cells and olive flounder (Paralichthys olivaceus). Fish Shellfish Immunol. 2012, 32, 459–463. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, K.H. The role of viral hemorrhagic septicemia virus (VHSV) NV gene in TNF-alpha- and VHSV infection-mediated NF-kappaB activation. Fish Shellfish Immunol. 2013, 34, 1315–1319. [Google Scholar] [CrossRef]
- Ammayappan, A.; Kurath, G.; Thompson, T.M.; Vakharia, V.N. A reverse genetics system for the Great Lakes strain of viral hemorrhagic septicemia virus: The NV gene is required for pathogenicity. Mar. Biotechnol. 2011, 13, 672–683. [Google Scholar] [CrossRef]
- Kesterson, S.P.; Ringiesn, J.; Vakharia, V.N.; Shepherd, B.S.; Leaman, D.W.; Malathi, K. Effect of the Viral Hemorrhagic Septicemia Virus Nonvirion Protein on Translation via PERK-eIF2alpha Pathway. Viruses 2020, 12, 499. [Google Scholar] [CrossRef]
- Collet, B.; Boudinot, P.; Benmansour, A.; Secombes, C.J. An Mx1 promoter-reporter system to study interferon pathways in rainbow trout. Dev. Comp. Immunol. 2004, 28, 793–801. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Gorgoglione, B.; Ringiesn, J.L.; Pham, L.H.; Shepherd, B.S.; Leaman, D.W. Comparative effects of Novirhabdovirus genes on modulating constitutive transcription and innate antiviral responses, in different teleost host cell types. Virol. J. 2020, 17, 110. [Google Scholar] [CrossRef]
- Faisal, M.; Shavalier, M.; Kim, R.K.; Millard, E.V.; Gunn, M.R.; Winters, A.D.; Schulz, C.A.; Eissa, A.; Thomas, M.V.; Wolgamood, M.; et al. Spread of the emerging viral hemorrhagic septicemia virus strain, genotype IVb, in Michigan, USA. Viruses 2012, 4, 734–760. [Google Scholar] [CrossRef]
- Liu, J.; HuangFu, W.C.; Kumar, K.G.; Qian, J.; Casey, J.P.; Hamanaka, R.B.; Grigoriadou, C.; Aldabe, R.; Diehl, J.A.; Fuchs, S.Y. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 2009, 5, 72–83. [Google Scholar] [CrossRef]
- Jordan, R.; Wang, L.; Graczyk, T.M.; Block, T.M.; Romano, P.R. Replication of a cytopathic strain of bovine viral diarrhea virus activates PERK and induces endoplasmic reticulum stress-mediated apoptosis of MDBK cells. J. Virol. 2002, 76, 9588–9599. [Google Scholar] [CrossRef]
- Echavarria-Consuegra, L.; Cook, G.M.; Busnadiego, I.; Lefevre, C.; Keep, S.; Brown, K.; Doyle, N.; Dowgier, G.; Franaszek, K.; Moore, N.A.; et al. Manipulation of the unfolded protein response: A pharmacological strategy against coronavirus infection. PLoS Pathog. 2021, 17, e1009644. [Google Scholar] [CrossRef]
- Ambrose, R.L.; Mackenzie, J.M. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 2011, 85, 2723–2732. [Google Scholar] [CrossRef]
- Lewy, T.G.; Offerdahl, D.K.; Grabowski, J.M.; Kellman, E.; Mlera, L.; Chiramel, A.; Bloom, M.E. PERK-Mediated Unfolded Protein Response Signaling Restricts Replication of the Tick-Borne Flavivirus Langat Virus. Viruses 2020, 12, 328. [Google Scholar] [CrossRef]
- Baltzis, D.; Qu, L.K.; Papadopoulou, S.; Blais, J.D.; Bell, J.C.; Sonenberg, N.; Koromilas, A.E. Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2alpha kinases PERK and PKR. J. Virol. 2004, 78, 12747–12761. [Google Scholar] [CrossRef]
- Nelson, E.V.; Schmidt, K.M.; Deflube, L.R.; Doganay, S.; Banadyga, L.; Olejnik, J.; Hume, A.J.; Ryabchikova, E.; Ebihara, H.; Kedersha, N.; et al. Ebola Virus Does Not Induce Stress Granule Formation during Infection and Sequesters Stress Granule Proteins within Viral Inclusions. J. Virol. 2016, 90, 7268–7284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, H.M.; Mohr, I. Defining the Role of Stress Granules in Innate Immune Suppression by the Herpes Simplex Virus 1 Endoribonuclease VHS. J. Virol. 2018, 92, e00829-18. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wu, S.; Kang, S.; Liao, J.; Zhang, L.; Xu, Z.; Chen, H.; Xu, L.; Zhang, X.; Qin, Q.; et al. Critical Roles of G3BP1 in Red-Spotted Grouper Nervous Necrosis Virus-Induced Stress Granule Formation and Viral Replication in Orange-Spotted Grouper (Epinephelus coioides). Front. Immunol. 2022, 13, 931534. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Burdeinick-Kerr, R.; Whelan, S.P. A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAs. Proc. Natl. Acad. Sci. USA 2013, 110, 324–329. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) |
|---|---|
| GFP se | ATGGTGAGCAAGGGCGAGGA |
| GFP as | TAGCGGCTGAAGCACTGCACGCC |
| VHSV Ia N qRT se | TTGATGAGACAGGTGTCAGAGG |
| VHSV Ia N qRT as | TTGGAGTTGTCATTGAGTCCAT |
| VHSV Ia P qRT se | GCTCCTGAGACGTATCAAGATG |
| VHSV Ia P qRT as | CATTTTCCTTTTGAGACTCCAG |
| VHSV Ia G qRT se | GGTGACTGTGACTATGAGGCAG |
| VHSV Ia G qRT as | CAACTTGTCCCCAAATATCATG |
| VHSV Ia M qRT se | TATGATCTTTGGAGAAACCAGC |
| VHSV Ia M qRT as | GTCACACTCCCATGTCTAATGG |
| VHSV Ia NV qRT se | GCGAGATGATCACACACAGACT |
| VHSV Ia NV qRT as | CCCTCAGATCATCTAGGATCCT |
| VHSV Ia L qRT se | AGAGAGCACATCAGGTACCAAG |
| VHSV Ia L qRT as | GCTCTGTGTCTTCAAAAGATGG |
| Fish IFN se | GATGCTGAGTTTGAGGACAAAGTC |
| Fish IFN as | GTTTCATGGCAGGTGATACACAGGA |
| Fish MX-1 se | ATTAACCTGGTTGTGGTGCCATGC |
| Fish MX-1 as | TACCACTGTCCCTTCAGTGCCTTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramnani, B.; Powell, S.; Shetty, A.G.; Manivannan, P.; Hibbard, B.R.; Leaman, D.W.; Malathi, K. Viral Hemorrhagic Septicemia Virus Activates Integrated Stress Response Pathway and Induces Stress Granules to Regulate Virus Replication. Viruses 2023, 15, 466. https://doi.org/10.3390/v15020466
Ramnani B, Powell S, Shetty AG, Manivannan P, Hibbard BR, Leaman DW, Malathi K. Viral Hemorrhagic Septicemia Virus Activates Integrated Stress Response Pathway and Induces Stress Granules to Regulate Virus Replication. Viruses. 2023; 15(2):466. https://doi.org/10.3390/v15020466
Chicago/Turabian StyleRamnani, Barkha, Shelby Powell, Adarsh G. Shetty, Praveen Manivannan, Brian R. Hibbard, Douglas W. Leaman, and Krishnamurthy Malathi. 2023. "Viral Hemorrhagic Septicemia Virus Activates Integrated Stress Response Pathway and Induces Stress Granules to Regulate Virus Replication" Viruses 15, no. 2: 466. https://doi.org/10.3390/v15020466
APA StyleRamnani, B., Powell, S., Shetty, A. G., Manivannan, P., Hibbard, B. R., Leaman, D. W., & Malathi, K. (2023). Viral Hemorrhagic Septicemia Virus Activates Integrated Stress Response Pathway and Induces Stress Granules to Regulate Virus Replication. Viruses, 15(2), 466. https://doi.org/10.3390/v15020466