
Dynamics of Early Establishment of SARS-CoV-2 VOC Omicron Lineages in Minas Gerais, Brazil
 , , , , , , , , , , , and add
Show full author list
, , , , , , , , , , , and add
Show full author list
{kind=link}
{kind=link}
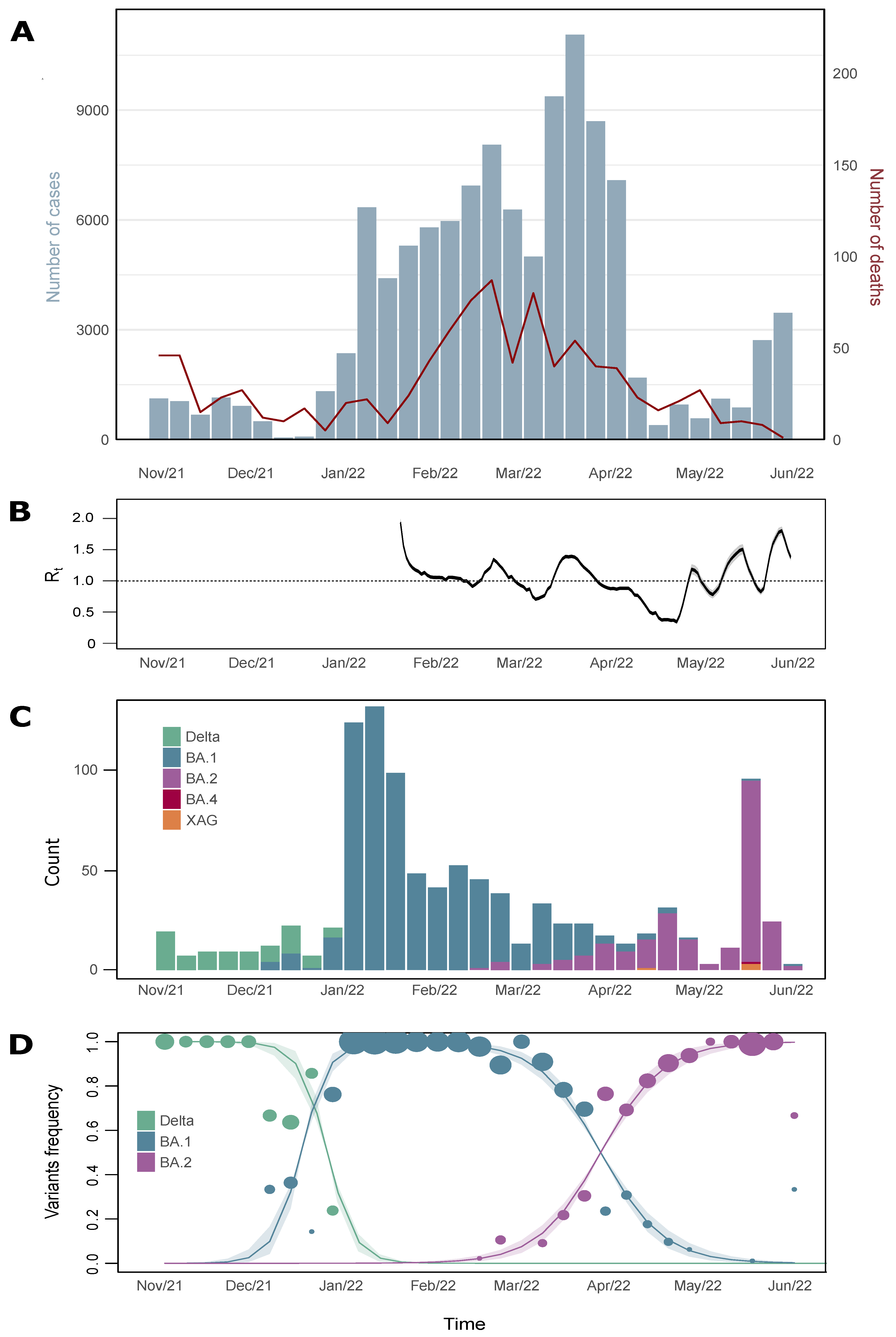
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and SARS-CoV-2 Diagnostic
2.2. Epidemiological Data
2.3. Multinomial Model
2.4. Genome Sequencing
2.5. Viral Genome Assembly and Classification
2.6. Phylogenetic Analyses
2.6.1. Maximum-Likelihood Phylogenetic Inference and Analysis of Temporal Signal
2.6.2. Bayesian Molecular Clock Analyses
2.6.3. Phylogeographic Inferences
3. Results
3.1. SARS-CoV-2 Omicron Replacement of Delta in MG
3.2. Phylogeographic Origins of VOC Omicron Lineages in Minas Gerais
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castro, M.C.; Kim, S.; Barberia, L.; Ribeiro, A.F.; Gurzenda, S.; Ribeiro, K.B.; Abbott, E.; Blossom, J.; Rache, B.; Singer, B.H. Spatiotemporal Pattern of COVID-19 Spread in Brazil. Science 2021, 372, 821–826. [Google Scholar] [CrossRef] [PubMed]
- de Jesus, J.G.; Sacchi, C.; da Silva Candido, D.; Claro, I.M.; Sales, F.C.S.; Manuli, E.R.; da Silva, D.B.B.; de Paiva, T.M.; Pinho, M.A.B.; de Oliveira Santos, K.C.; et al. Importation and Early Local Transmission of COVID-19 in Brazil, 2020. Rev. Inst. Med. Trop. Sao Paulo 2020, 62, e30. [Google Scholar] [CrossRef] [PubMed]
- Candido, D.S.; Claro, I.M.; de Jesus, J.G.; Souza, W.M.; Moreira, F.R.R.; Dellicour, S.; Mellan, T.A.; du Plessis, L.; Pereira, R.H.M.; Sales, F.C.S.; et al. Evolution and Epidemic Spread of SARS-CoV-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef]
- Noureddine, F.Y.; Chakkour, M.; El Roz, A.; Reda, J.; Al Sahily, R.; Assi, A.; Joma, M.; Salami, H.; Hashem, S.J.; Harb, B.; et al. The Emergence of SARS-CoV-2 Variant(s) and Its Impact on the Prevalence of COVID-19 Cases in the Nabatieh Region, Lebanon. Med. Sci. 2021, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; da S. Candido, D.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, G. Sequence Analysis of the Emerging SARS-CoV-2 Variant Omicron in South Africa. J. Med. Virol. 2022, 94, 1728–1733. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain That Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Tsang, A.K.; Cheng, P.K.; Mak, G.C.; Leung, P.K.; Yip, P.C.; Lam, E.T.; Ng, K.H.; Chan, R.C. Unusual High Number of Spike Protein Mutations for the SARS-CoV-2 Strains Detected in Hong Kong. J. Clin. Virol. 2022, 148, 105081. [Google Scholar] [CrossRef]
- Torjesen, I. Covid-19: Omicron May Be More Transmissible than Other Variants and Partly Resistant to Existing Vaccines, Scientists Fear. BMJ 2021, 375, n2943. [Google Scholar] [CrossRef]
- The R Project for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 21 December 2022).
- Cori, A.; Ferguson, N.M.; Fraser, C.; Cauchemez, S. A New Framework and Software to Estimate Time-Varying Reproduction Numbers during Epidemics. Am. J. Epidemiol. 2013, 178, 1505–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, L. ggplot2: Elegant Graphics for Data Analysis by WICKHAM, H. Biometrics 2011, 67, 678–679. [Google Scholar] [CrossRef]
- Yu, G. Using Ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Campbell, F.; Archer, B.; Laurenson-Schafer, H.; Jinnai, Y.; Konings, F.; Batra, N.; Pavlin, B.; Vandemaele, K.; Van Kerkhove, M.D.; Jombart, T.; et al. Increased Transmissibility and Global Spread of SARS-CoV-2 Variants of Concern as at June 2021. Euro Surveill. 2021, 26, 2100509. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Gelman, A.; Hoffman, M.D.; Lee, D.; Goodrich, B.; Betancourt, M.; Brubaker, M.; Guo, J.; Li, P.; Riddell, A. Stan: A Probabilistic Programming Language. J. Stat. Softw. 2017, 76, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed, N.E.; Vlková, M.; Faisal, M.B.; Silander, O.K. Rapid and Inexpensive Whole-Genome Sequencing of SARS-CoV-2 Using 1200 Bp Tiled Amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc. 2020, 5, bpaa014. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap and Miniasm: Fast Mapping and de Novo Assembly for Noisy Long Sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Fonseca, P.L.C.; Moreira, F.R.R.; de Souza, R.M.; Guimarães, N.R.; Carvalho, N.O.; Adelino, T.E.R.; Alves, H.J.; Alvim, L.B.; Candido, D.S.; Coelho, H.P.; et al. Tracking the Turnover of SARS-CoV-2 VOCs Gamma to Delta in a Brazilian State (Minas Gerais) with a High-Vaccination Status. Virus Evol. 2022, 8, veac064. [Google Scholar] [CrossRef] [PubMed]
- de S. Silva, T.d.S.; Salvato, R.S.; Gregianini, T.S.; Gomes, I.A.; Pereira, E.C.; de Oliveira, E.; de Menezes, A.L.; Barcellos, R.B.; Godinho, F.M.; Riediger, I.; et al. Molecular Characterization of a New SARS-CoV-2 Recombinant Cluster XAG Identified in Brazil. Front. Med. 2022, 9, 1008600. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. Maximum Likelihood Phylogenetic Estimation from DNA Sequences with Variable Rates over Sites: Approximate Methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didelot, X.; Siveroni, I.; Volz, E.M. Additive Uncorrelated Relaxed Clock Models for the Dating of Genomic Epidemiology Phylogenies. Mol. Biol. Evol. 2021, 38, 307–317. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Luna, N.; Muñoz, M.; Ramírez, A.L.; Patiño, L.H.; Castañeda, S.A.; Ballesteros, N.; Ramírez, J.D. Genomic Diversity of SARS-CoV-2 Omicron Variant in South American Countries. Viruses 2022, 14, 1234. [Google Scholar] [CrossRef]
- Gutierrez, B.; Márquez, S.; Prado-Vivar, B.; Becerra-Wong, M.; Guadalupe, J.J.; Candido, D.D.S.; Fernandez-Cadena, J.C.; Morey-Leon, G.; Armas-Gonzalez, R.; Andrade-Molina, D.M.; et al. Genomic Epidemiology of SARS-CoV-2 Transmission Lineages in Ecuador. Virus Evol. 2021, 7, veab051. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, M.; Yano, T.-A.; Kishino, H. A New Molecular Clock of Mitochondrial DNA and the Evolution of Hominoids. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 1984, 60, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian Population Dynamics Inference: A Coalescent-Based Model for Multiple Loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minin, V.N.; Suchard, M.A. Counting Labeled Transitions in Continuous-Time Markov Models of Evolution. J. Math. Biol. 2007, 56, 391–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Krüger, N.; Schulz, S.; Cossmann, A.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Moldenhauer, A.-S.; Winkler, M.S.; et al. The Omicron Variant Is Highly Resistant against Antibody-Mediated Neutralization: Implications for Control of the COVID-19 Pandemic. Cell 2022, 185, 447–456.e11. [Google Scholar] [CrossRef]
- Syed, A.M.; Ciling, A.; Taha, T.Y.; Chen, I.P.; Khalid, M.M.; Sreekumar, B.; Chen, P.-Y.; Kumar, G.R.; Suryawanshi, R.; Silva, I.; et al. Omicron Mutations Enhance Infectivity and Reduce Antibody Neutralization of SARS-CoV-2 Virus-like Particles. Proc. Natl. Acad. Sci. USA 2022, 119, e2200592119. [Google Scholar] [CrossRef] [PubMed]
- Adamoski, D.; de Baura, V.A.; Rodrigues, A.C.; Royer, C.A.; Aoki, M.N.; Tschá, M.K.; Bonatto, A.C.; Wassem, R.; Nogueira, M.B.; Raboni, S.M.; et al. SARS-CoV-2 Delta and Omicron Variants Surge in Curitiba, Southern Brazil, and Its Impact on Overall COVID-19 Lethality. Viruses 2022, 14, 809. [Google Scholar] [CrossRef]
- Duong, B.V.; Larpruenrudee, P.; Fang, T.; Hossain, S.I.; Saha, S.C.; Gu, Y.; Islam, M.S. Is the SARS CoV-2 Omicron Variant Deadlier and More Transmissible Than Delta Variant? Int. J. Environ. Res. Public Health 2022, 19, 4586. [Google Scholar] [CrossRef]
- Mahase, E. Omicron Sub-Lineage BA.2 May Have “Substantial Growth Advantage,” UKHSA reports. BMJ 2022, 376, o263. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron Lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef]
- Queiroz, D.C.; Carobin, N.V.; de Araújo e Santos, L.C.G.; Fonseca, P.L.C.; Braga-Paz, I.L.; Dias, R.C.; Ferreira, J.G.G.; Freitas, T.R.; Menezes, D.; Nolasco, S.C.V.M.; et al. SARS-CoV-2 Omicron BA.1, BA.2, and XAG Identification during Routine Surveillance on a University Campus in Belo Horizonte, Brazil, 2022. Braz. J. Microbiol. 2022, 53, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Slavov, S.N.; Fonseca, V.; Wilkinson, E.; Tegally, H.; Patané, J.S.L.; Viala, V.L.; San, E.J.; Rodrigues, E.S.; Santos, E.V.; et al. Genomic Epidemiology of the SARS-CoV-2 Epidemic in Brazil. Nat. Microbiol. 2022, 7, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Xavier, J.; Giovanetti, M.; Adelino, T.; Fonseca, V.; Barbosa da Costa, A.V.; Ribeiro, A.A.; Felicio, K.N.; Duarte, C.G.; Ferreira Silva, M.V.; Salgado, Á.; et al. The Ongoing COVID-19 Epidemic in Minas Gerais, Brazil: Insights from Epidemiological Data and SARS-CoV-2 Whole Genome Sequencing. Emerg. Microbes Infect. 2020, 9, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Moreira, F.R.R.; D’arc, M.; Mariani, D.; Herlinger, A.L.; Schiffler, F.B.; Rossi, Á.D.; de Carvalho Leitão, I.; dos Santos Miranda, T.; Cosentino, M.A.C.; de Paula Tôrres, M.C.; et al. Epidemiological Dynamics of SARS-CoV-2 VOC Gamma in Rio de Janeiro, Brazil. Virus Evol. 2021, 7, veab087. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Wegrzyn, R.D.; Appiah, G.D.; Morfino, R.; Milford, S.R.; Walker, A.T.; Ernst, E.T.; Darrow, W.W.; Li, S.L.; Robison, K.; MacCannell, D.; et al. Early Detection of SARS-CoV-2 Variants Using Traveler-Based Genomic Surveillance at Four US Airports, September 2021–January 2022. Clin. Infect. Dis. 2022, ciac461. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Menezes, M.T.; Moreira, F.R.R.; Whittaker, C.; Santos, F.M.; Queiroz, D.C.; Geddes, V.; Fonseca, P.L.C.; de Jesus, J.G.; Mendes-Oliveira, F.; Reis-Souza, V.; et al. Dynamics of Early Establishment of SARS-CoV-2 VOC Omicron Lineages in Minas Gerais, Brazil. Viruses 2023, 15, 585. https://doi.org/10.3390/v15020585
de Menezes MT, Moreira FRR, Whittaker C, Santos FM, Queiroz DC, Geddes V, Fonseca PLC, de Jesus JG, Mendes-Oliveira F, Reis-Souza V, et al. Dynamics of Early Establishment of SARS-CoV-2 VOC Omicron Lineages in Minas Gerais, Brazil. Viruses. 2023; 15(2):585. https://doi.org/10.3390/v15020585
Chicago/Turabian Stylede Menezes, Mariane Talon, Filipe Romero Rebello Moreira, Charles Whittaker, Franciele Martins Santos, Daniel Costa Queiroz, Victor Geddes, Paula Luize Camargos Fonseca, Jaqueline Góes de Jesus, Franciane Mendes-Oliveira, Valquíria Reis-Souza, and et al. 2023. "Dynamics of Early Establishment of SARS-CoV-2 VOC Omicron Lineages in Minas Gerais, Brazil" Viruses 15, no. 2: 585. https://doi.org/10.3390/v15020585
APA Stylede Menezes, M. T., Moreira, F. R. R., Whittaker, C., Santos, F. M., Queiroz, D. C., Geddes, V., Fonseca, P. L. C., de Jesus, J. G., Mendes-Oliveira, F., Reis-Souza, V., Santos, B., Zauli, D. A. G., de Lima, A. B., de Brito Mendonça, C., Alvim, L. B., do Prado Silva, J., Malta, F. S. V., de Souza Ferreira, A. C., Faria, N. R., ... Aguiar, R. S. (2023). Dynamics of Early Establishment of SARS-CoV-2 VOC Omicron Lineages in Minas Gerais, Brazil. Viruses, 15(2), 585. https://doi.org/10.3390/v15020585