
Epstein–Barr Virus History and Pathogenesis



Abstract
:1. Introduction
2. EBV History
3. Biology and the Potent Transforming Ability of Oncogenic EBV
4. Pathogenesis Associated with EBV Latent and Lytic Infection
4.1. EBV Latent Infection. Contribution of EBV Latent Antigens to the Oncogenic Phenotype Remains the Subject of Intense Study
4.1.1. The EBV-Encoded Nuclear Antigens (Summarized in Inset 1)

4.1.2. EBV-Encoded Latent Membrane Antigens (Summarized in Inset 2)

4.1.3. EBV-Encoded Functional Small Non-Coding RNAs
4.2. EBV Lytic Genes and Their Associated Functions
5. EBV-Associated Malignances
5.1. EBV-Associated Lymphomas
5.1.1. EBV-Associated B-Cell Lymphomas
Burkitt’s Lymphoma
Hodgkin Lymphoma
EBV-Positive Diffuse Large B-Cell Lymphoma
5.1.2. EBV-Associated NK-Cell and T-Cell Lymphomas
Extranodal Natural Killer/T-Cell Lymphoma
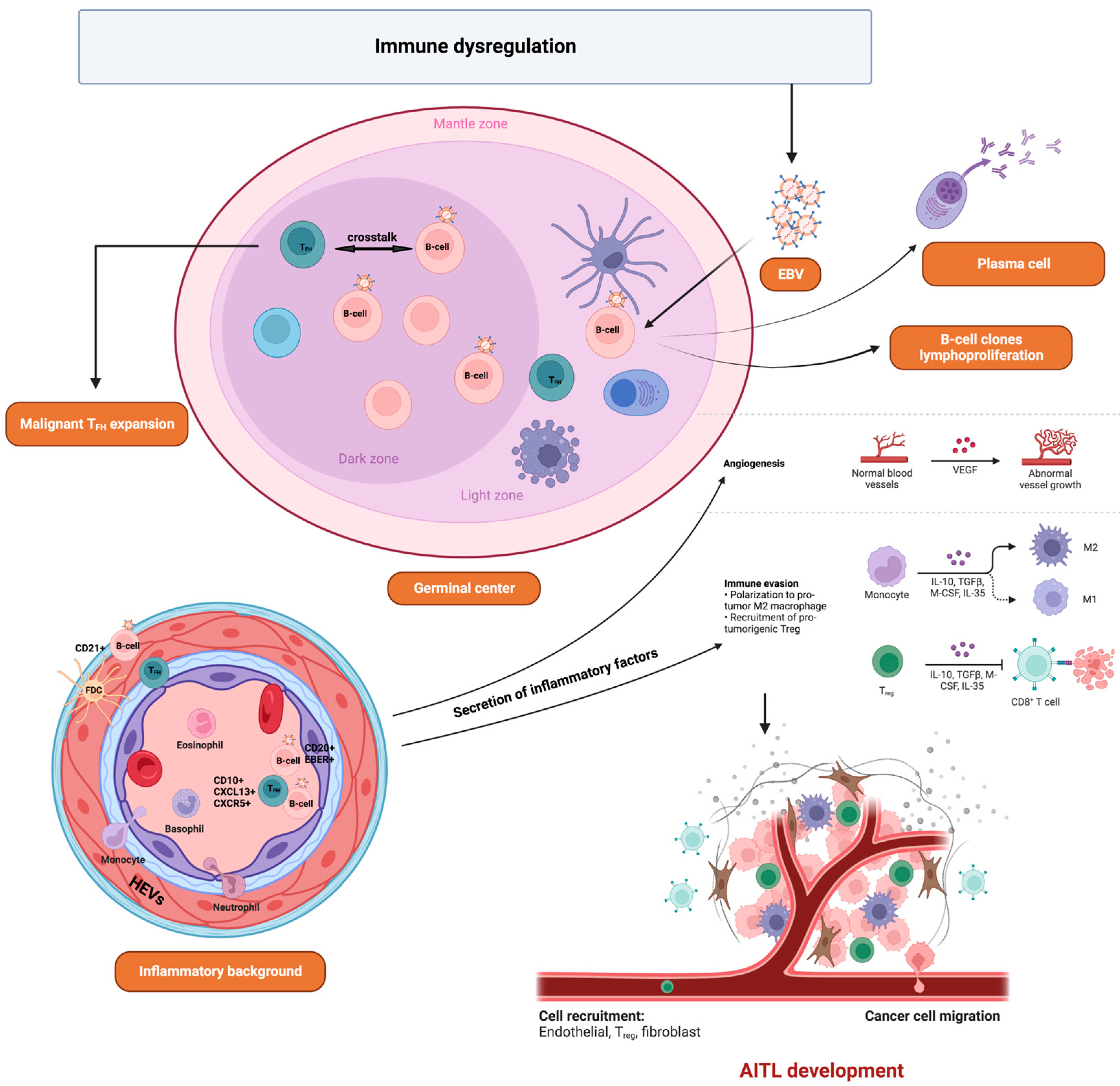
Angioimmunoblastic T-Cell Lymphoma
5.2. EBV-Associated Epithelial Carcinomas
5.2.1. Nasopharyngeal Carcinoma
5.2.2. Gastric Cancer
6. Summary and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, J.I. Epstein-barr virus infection. N. Engl. J. Med. 2000, 343, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, K.; Sullivan, J.L. Infectious mononucleosis. N. Engl. J. Med. 2010, 362, 1993–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the epstein-barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the virus. Curr. Top Microbiol. Immunol. 2015, 390 Pt 1, 151–209. [Google Scholar]
- Tonoyan, L.; Vincent-Bugnas, S.; Olivieri, C.V.; Doglio, A. New viral facets in oral diseases: The EBV paradox. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Robinson, W.H.; Steinman, L. Epstein-barr virus and multiple sclerosis. Science 2022, 375, 264–265. [Google Scholar] [CrossRef]
- Houen, G.; Trier, N.H. Epstein-barr virus and systemic autoimmune diseases. Front. Immunol. 2020, 11, 587380. [Google Scholar] [CrossRef]
- Farrell, P.J. Epstein-barr virus and cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- de Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Old, L.J.; Boyse, E.A.; Oettgen, H.F.; Harven, E.D.; Geering, G.; Williamson, B.; Clifford, P. Precipitating antibody in human serum to an antigen present in cultured burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. USA 1966, 56, 1699–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.A.; Henle, G. Indirect immunofluorescence tests with sera from African children and cultured Burkitt lymphoma cells. J. Bacteriol. 1966, 92, 275–276. [Google Scholar] [CrossRef] [Green Version]
- Henle, G.; Henle, W.; Diehl, V. Relation of burkitt’s tumor-associated herpes-ytpe virus to infectious mononucleosis. Proc. Natl. Acad. Sci. USA 1968, 59, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- zur Hausen, H.; Schulte-Holthausen, H.; Klein, G.; Henle, W.; Henle, G.; Clifford, P.; Santesson, L. EBV DNA in biopsies of burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 1970, 228, 1056–1058. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.; zur Hausen, H.; Becker, V. EB viral genomes in epithelial nasopharyngeal carcinoma cells. Nat. New Biol. 1973, 244, 245–247. [Google Scholar] [PubMed] [Green Version]
- Ziegler, J.L.; Drew, W.L.; Miner, R.C.; Mintz, L.; Rosenbaum, E.; Gershow, J.; Lennette, E.T.; Greenspan, J.; Shillitoe, E.; Beckstead, J.; et al. Outbreak of burkitt’s-like lymphoma in homosexual men. Lancet 1982, 2, 631–633. [Google Scholar] [CrossRef]
- Greenspan, J.S.; Greenspan, D.; Lennette, E.T.; Abrams, D.I.; Conant, M.A.; Petersen, V.; Freese, U.K. Replication of epstein-barr virus within the epithelial cells of oral “hairy” leukoplakia, an AIDS-associated lesion. N. Engl. J. Med. 1985, 313, 1564–1571. [Google Scholar] [CrossRef]
- Nagington, J.; Gray, J. Cyclosporin A immunosuppression, epstein-barr antibody, and lymphoma. Lancet 1980, 1, 536–537. [Google Scholar] [CrossRef]
- Weiss, L.M.; Movahed, L.A.; Warnke, R.A.; Sklar, J. Detection of epstein-barr viral genomes in reed-sternberg cells of hodgkin’s disease. N. Engl. J. Med. 1989, 320, 502–506. [Google Scholar] [CrossRef]
- Jones, J.F.; Shurin, S.; Abramowsky, C.; Tubbs, R.R.; Sciotto, C.G.; Wahl, R.; Sands, J.; Gottman, D.; Katz, B.Z.; Sklar, J. T-cell lymphomas containing Epstein-Barr viral DNA in patients with chronic epstein-barr virus infections. N. Engl. J. Med. 1988, 318, 733–741. [Google Scholar] [CrossRef]
- Iwatsuki, K.; Miyake, T.; Hirai, Y.; Yamamoto, T. Hydroa vacciniforme: A distinctive form of epstein-barr virus-associated T-cell lymphoproliferative disorders. Eur. J. Dermatol. 2019, 29, 21–28. [Google Scholar] [PubMed]
- Hue, S.S.; Oon, M.L.; Wang, S.; Tan, S.Y.; Ng, S.B. Epstein-barr virus-associated T- and NK-cell lymphoproliferative diseases: An update and diagnostic approach. Pathology 2020, 52, 111–127. [Google Scholar] [CrossRef]
- Shibata, D.; Weiss, L.M. Epstein-barr virus-associated gastric adenocarcinoma. Am. J. Pathol. 1992, 140, 769–774. [Google Scholar] [PubMed]
- Bonnet, M.; Guinebretiere, J.M.; Kremmer, E.; Grunewald, V.; Benhamou, E.; Contesso, G.; Joab, I. Detection of epstein-barr virus in invasive breast cancers. J. Natl. Cancer Inst. 1999, 91, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, Y.; Mizugaki, Y.; Uchida, T.; Torii, T.; Imai, S.; Makuuchi, M.; Takada, K. Detection of epstein-barr virus (EBV) in hepatocellular carcinoma tissue: A novel EBV latency characterized by the absence of EBV-encoded small RNA expression. Virology 1999, 256, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, D.H. Biology and disease associations of epstein-barr virus. Philos. Trans. R Soc. Lond B Biol. Sci. 2001, 356, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. Epstein-barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Dunmire, S.K.; Grimm, J.M.; Schmeling, D.O.; Balfour, H.H.J.; Hogquist, K.A. The incubation period of primary epstein-barr virus infection: Viral dynamics and immunologic events. PLoS Pathog. 2015, 11, e1005286. [Google Scholar] [CrossRef]
- Tanner, J.; Weis, J.; Fearon, D.; Whang, Y.; Kieff, E. Epstein-barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 1987, 50, 203–213. [Google Scholar] [CrossRef]
- Savard, M.; Belanger, C.; Tardif, M.; Gourde, P.; Flamand, L.; Gosselin, J. Infection of primary human monocytes by epstein-barr virus. J. Virol. 2000, 74, 2612–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrei, G.; Trompet, E.; Snoeck, R. Novel therapeutics for epstein(-)barr virus. Molecules 2019, 24, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, H.C.; Mehta, D.; Lu, J.; El-Naccache, D.; Shukla, S.K.; Kovacsics, C.; Kolson, D.; Robertson, E.S. Gammaherpesvirus infection of human neuronal cells. mBio 2015, 6, e01844-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutt-Fletcher, L.M. Epstein-Barr virus entry. J. Virol. 2007, 81, 7825–7832. [Google Scholar] [CrossRef] [Green Version]
- Sixbey, J.W.; Vesterinen, E.H.; Nedrud, J.G.; Raab-Traub, N.; Walton, L.A.; Pagano, J.S. Replication of epstein-barr virus in human epithelial cells infected in vitro. Nature 1983, 306, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.X.; Young, L.S.; Niedobitek, G.; Dawson, C.W.; Birkenbach, M.; Wang, F.; Rickinson, A.B. Epstein-barr virus infection and replication in a human epithelial cell system. Nature 1992, 356, 347–350. [Google Scholar] [CrossRef]
- Fox, C.P.; Shannon-Lowe, C.; Rowe, M. Deciphering the role of epstein-barr virus in the pathogenesis of T and NK cell lymphoproliferations. Herpesviridae 2011, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Henle, W.; Diehl, V.; Kohn, G.; Hausen, H.; Henle, G. Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated Burkitt cells. Science 1967, 157, 1064–1065. [Google Scholar] [CrossRef]
- Diehl, V.; Henle, G.; Henle, W.; Kohn, G. Demonstration of a herpes group virus in cultures of peripheral leukocytes from patients with infectious mononucleosis. J. Virol. 1968, 2, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Baer, R.; Bankier, A.T.; Biggin, M.D.; Deininger, P.L.; Farrell, P.J.; Gibson, T.J.; Hatfull, G.; Hudson, G.S.; Satchwell, S.C.; Seguin, C.; et al. DNA sequence and expression of the B95-8 epstein-barr virus genome. Nature 1984, 310, 207–211. [Google Scholar] [CrossRef]
- Dambaugh, T.; Hennessy, K.; Chamnankit, L.; Kieff, E. U2 region of epstein-barr virus DNA may encode epstein-barr nuclear antigen 2. Proc. Natl. Acad. Sci. USA 1984, 81, 7632–7636. [Google Scholar] [CrossRef] [Green Version]
- Sample, J.; Young, L.; Martin, B.; Chatman, T.; Kieff, E.; Rickinson, A.; Kieff, E. Epstein-barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J. Virol. 1990, 64, 4084–4092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrand, J.R.; Young, L.S.; Tugwood, J.D. Two families of sequences in the small RNA-encoding region of epstein-barr virus (EBV) correlate with EBV types A and B. J. Virol. 1989, 63, 983–986. [Google Scholar] [CrossRef] [Green Version]
- Zimber, U.; Adldinger, H.K.; Lenoir, G.M.; Vuillaume, M.; Knebel-Doeberitz, M.V.; Laux, G.; Desgranges, C.; Wittmann, P.; Freese, U.K.; Schneider, U.; et al. Geographical prevalence of two types of epstein-barr virus. Virology 1986, 154, 56–66. [Google Scholar] [CrossRef]
- Kuppers, R. B cells under influence: Transformation of B cells by epstein-barr virus. Nat. Rev. Immunol. 2003, 3, 801–812. [Google Scholar] [CrossRef]
- Hochberg, D.; Middeldorp, J.M.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Demonstration of the burkitt’s lymphoma epstein-barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 239–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, G. Viral latency and transformation: The strategy of epstein-barr virus. Cell 1989, 58, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.A.; Lear, A.L.; Young, L.S.; Rickinson, A.B. Transcripts from the epstein-barr virus bamHI A fragment are detectable in all three forms of virus latency. J. Virol. 1993, 67, 3182–3190. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Robertson, E.S. Mechanisms of B-cell oncogenesis induced by epstein-barr virus. J. Virol. 2019, 93, e00238-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munz, C. Latency and lytic replication in epstein-barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.K.; Gulley, M.L.; Feng, W.H.; Delecluse, H.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of epstein-barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frappier, L. Epstein-barr virus: Current questions and challenges. Tumour. Virus Res. 2021, 12, 200218. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.L.; Warren, N.; Sugden, B. Stable replication of plasmids derived from epstein-barr virus in various mammalian cells. Nature 1985, 313, 812–815. [Google Scholar] [CrossRef]
- Westhoff Smith, D.; Sugden, B. Potential cellular functions of epstein-barr nuclear antigen 1 (EBNA1) of epstein-barr virus. Viruses 2013, 5, 226–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, G.; Komano, J.; Sugden, B. Epstein-barr virus provides a survival factor to burkitt’s lymphomas. Proc. Natl. Acad. Sci. USA 2003, 100, 14269–14274. [Google Scholar] [CrossRef] [Green Version]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The epstein-barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.B.; Bell, J.L.; Levine, A.J. Expression of epstein-barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. EMBO J. 1996, 15, 3117–3126. [Google Scholar] [CrossRef]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of antigen processing by the internal repeat region of the epstein-barr virus nuclear antigen-1. Nature 1995, 375, 685–688. [Google Scholar] [CrossRef]
- Levitskaya, J.; Sharipo, A.; Leonchiks, A.; Ciechanover, A.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 1997, 94, 12616–12621. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Saridakis, V.; Sarkari, F.; Duan, S.; Wu, T.; Arrowsmith, C.H.; Frappier, L. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 2006, 13, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R.; Johannsen, E.; Tong, X.; Yalamanchili, R.; Kieff, E. The epstein-barr virus nuclear antigen 2 transactivator is directed to response elements by the j kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 1994, 91, 7568–7572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.J.; Hayward, S.D. Masking of the CBF1/RBPJ kappa transcriptional repression domain by epstein-barr virus EBNA2. Science 1995, 268, 560–563. [Google Scholar] [CrossRef]
- Zhao, B.; Maruo, S.; Cooper, A.; R. Chase, M.; Johannsen, E.; Kieff, E.; Cahir-McFarland, E. RNAs induced by epstein-barr virus nuclear antigen 2 in lymphoblastoid cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 1900–1905. [Google Scholar] [CrossRef] [Green Version]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef]
- Hong, T.; Parameswaran, S.; Donmez, O.A.; Miller, D.; Forney, C.; Lape, M.; Saint Just Ribeiro, M.; Liang, J.; Edsall, L.E.; Magnusen, A.F.; et al. Epstein-barr virus nuclear antigen 2 extensively rewires the human chromatin landscape at autoimmune risk loci. Genome. Res. 2021, 31, 2185–2198. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Matsuno, K.; Fortini, M.E. Notch signaling. Science 1995, 268, 225–232. [Google Scholar] [CrossRef]
- Sakai, T.; Taniguchi, Y.; Tamura, K.; Minoguchi, S.; Fukuhara, T.; Strobl, L.J.; Zimber-Strobl, U.; Bornkamm, G.W.; Honjo, T. Functional replacement of the intracellular region of the Notch1 receptor by Epstein-Barr virus nuclear antigen 2. J. Virol. 1998, 72, 6034–6039. [Google Scholar] [CrossRef] [Green Version]
- Robertson, E.S.; Lin, J.; Kieff, E. The amino-terminal domains of epstein-barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ(kappa). J. Virol. 1996, 70, 3068–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radkov, S.A.; Touitou, R.; Brehm, A.; Rowe, M.; West, M.; Kouzarides, T.; Allday, M.J. Epstein-barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J. Virol. 1999, 73, 5688–5697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymula, A.; Palermo, R.D.; Bayoumy, A.; Groves, I.J.; Ba Abdullah, M.; Holder, B.; White, R.E. Epstein-barr virus nuclear antigen EBNA-LP is essential for transforming naive B cells, and facilitates recruitment of transcription factors to the viral genome. PLoS Pathog. 2018, 14, e1006890. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I.; Wang, F.; Mannick, J.; Kieff, E. Epstein-barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc. Natl. Acad. Sci. USA 1989, 86, 9558–9562. [Google Scholar] [CrossRef] [Green Version]
- Tomkinson, B.; Robertson, E.; Kieff, E. Epstein-barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 1993, 67, 2014–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tursiella, M.L.; Bowman, E.R.; Wanzeck, K.C.; Throm, R.E.; Liao, J.; Zhu, J.; Sample, C.E. Epstein-barr virus nuclear antigen 3A promotes cellular proliferation by repression of the cyclin-dependent kinase inhibitor p21WAF1/CIP1. PLoS Pathog. 2014, 10, e1004415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, J.S.; Robertson, E.S. Epstein-barr virus nuclear antigen 3C regulates cyclin A/p27 complexes and enhances cyclin A-dependent kinase activity. J. Virol. 2004, 78, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Ghosh Roy, S.; Bose, P.; Saha, A. Role of EBNA-3 family proteins in EBV associated B-cell lymphomagenesis. Front. Microbiol. 2016, 7, 457. [Google Scholar] [CrossRef] [Green Version]
- Price, A.M.; Dai, J.; Bazot, Q.; Patel, L.; Nikitin, P.A.; Djavadian, R.; Winter, P.S.; Salinas, C.A.; Barry, A.P.; Wood, K.C.; et al. Epstein-barr virus ensures B cell survival by uniquely modulating apoptosis at early and late times after infection. Elife 2017, 6, e22509. [Google Scholar] [CrossRef]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene bim. PLoS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef] [Green Version]
- Styles, C.T.; Bazot, Q.; Parker, G.A.; White, R.E.; Paschos, K.; Allday, M.J. EBV epigenetically suppresses the B cell-to-plasma cell differentiation pathway while establishing long-term latency. PLoS Biol. 2017, 15, e2001992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Styles, C.T.; Paschos, K.; White, R.E.; Farrell, P.J. The cooperative functions of the EBNA3 proteins are central to EBV persistence and latency. Pathogens 2018, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gain, C.; Malik, S.; Bhattacharjee, S.; Ghosh, A.; Robertson, E.S.; Das, B.B.; Saha, A. Proteasomal inhibition triggers viral oncoprotein degradation via autophagy-lysosomal pathway. PLoS Pathog. 2020, 16, e1008105. [Google Scholar] [CrossRef] [Green Version]
- Kieser, A.; Sterz, K.R. The latent membrane protein 1 (LMP1). Curr. Top Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar]
- Eliopoulos, A.G.; Stack, M.; Dawson, C.W.; Kaye, K.M.; Hodgkin, L.; Sihota, S.; Rowe, M.; Young, L.S. Epstein-barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-kappaB pathway involving TNF receptor-associated factors. Oncogene 1997, 14, 2899–2916. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, K.; Das, P.; Chattopadhyay, N.R.; Mal, S.; Choudhuri, T. The interplay between epstein-bar virus (EBV) with the p53 and its homologs during EBV associated malignancies. Heliyon 2019, 5, e02624. [Google Scholar] [CrossRef]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-barr virus LMP1-mediated oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef] [Green Version]
- Chabay, P. Advances in the pathogenesis of EBV-associated diffuse large B cell lymphoma. Cancers 2021, 13, 2717. [Google Scholar] [CrossRef]
- Miller, W.E.; Mosialos, G.; Kieff, E.; Raab-Traub, N. Epstein-barr virus LMP1 induction of the epidermal growth factor receptor is mediated through a TRAF signaling pathway distinct from NF-kappaB activation. J. Virol. 1997, 71, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Eliopoulos, A.G.; Young, L.S. LMP1 structure and signal transduction. Semin. Cancer Biol. 2001, 11, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Sato, H.; Furukawa, M.; Pagano, J.S. The expression of matrix metalloproteinase 9 is enhanced by epstein-barr virus latent membrane protein 1. Proc. Natl. Acad. Sci. USA 1998, 95, 3621–3626. [Google Scholar] [CrossRef] [Green Version]
- Aravinth, S.P.; Rajendran, S.; Li, Y.; Wu, M.; Yi Wong, A.H.; Schwarz, H. Epstein-barr virus-encoded LMP1 induces ectopic CD137 expression on hodgkin and reed-sternberg cells via the PI3K-AKT-mTOR pathway. Leuk Lymphoma 2019, 60, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.W.; Wang, H.; Zhang, W.W.; Wang, J.H.; Liu, W.J.; Xia, Z.J.; Huang, H.Q.; Jiang, W.Q.; Zhang, Y.J.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-kappaB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longnecker, R.; Kieff, E. A second epstein-barr virus membrane protein (LMP2) is expressed in latent infection and colocalizes with LMP1. J. Virol. 1990, 64, 2319–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longnecker, R. Epstein-barr virus latency: LMP2, a regulator or means for epstein-barr virus persistence? Adv. Cancer Res. 2000, 79, 175–200. [Google Scholar] [PubMed]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Casola, S.; Otipoby, K.L.; Alimzhanov, M.; Humme, S.; Uyttersprot, N.; Kutok, J.L.; Carroll, M.C.; Rajewsky, K. B cell receptor signal strength determines B cell fate. Nat. Immunol. 2004, 5, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Fish, K.; Comoglio, F.; Shaffer, A.L., III; Ji, Y.; Pan, K.T.; Scheich, S.; Oellerich, A.; Doebele, C.; Ikeda, M.; Schaller, S.J.; et al. Rewiring of B cell receptor signaling by epstein-barr virus LMP2A. Proc. Natl. Acad. Sci. USA 2020, 117, 26318–26327. [Google Scholar] [CrossRef]
- Scholle, F.; Bendt, K.M.; Raab-Traub, N. Epstein-barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J. Virol. 2000, 74, 10681–10689. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.M.; Stewart, S.E.; Wei, W.; Woodman, C.B.; O’Neil, J.D.; Dawson, C.W.; Young, L.S. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene 2009, 28, 3903–3914. [Google Scholar] [CrossRef] [Green Version]
- Rovedo, M.; Longnecker, R. Epstein-barr virus latent membrane protein 2B (LMP2B) modulates LMP2A activity. J. Virol. 2007, 81, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Rechsteiner, M.P.; Berger, C.; Zauner, L.; Sigrist, J.A.; Weber, M.; Longnecker, R.; Bernasconi, M.; Nadal, D. Latent membrane protein 2B regulates susceptibility to induction of lytic epstein-barr virus infection. J. Virol. 2008, 82, 1739–1747. [Google Scholar] [CrossRef] [Green Version]
- Takada, K.; Nanbo, A. The role of EBERs in oncogenesis. Semin. Cancer Biol. 2001, 11, 461–467. [Google Scholar] [CrossRef]
- Niller, H.H.; Salamon, D.; Ilg, K.; Koroknai, A.; Banati, F.; Bauml, G.; Rucker, O.; Schwarzmann, F.; Wolf, H.; Minarovits, J. The in vivo binding site for oncoprotein c-Myc in the promoter for epstein-barr virus (EBV) encoding RNA (EBER) 1 suggests a specific role for EBV in lymphomagenesis. Med. Sci. Monit. 2003, 9, HY1-9. [Google Scholar]
- Komano, J.; Maruo, S.; Kurozumi, K.; Oda, T.; Takada, K. Oncogenic role of epstein-barr virus-encoded RNAs in burkitt’s lymphoma cell line Akata. J. Virol. 1999, 73, 9827–9831. [Google Scholar] [CrossRef] [Green Version]
- Ruf, I.K.; Rhyne, P.W.; Yang, C.; Cleveland, J.L.; Sample, J.T. Epstein-barr virus small RNAs potentiate tumorigenicity of burkitt lymphoma cells independently of an effect on apoptosis. J. Virol. 2000, 74, 10223–10228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.X.; de Turenne-Tessier, M.; Decaussin, G.; Benet, G.; Ooka, T. Establishment of a monkey kidney epithelial cell line with the BARF1 open reading frame from epstein-barr virus. Oncogene 1997, 14, 3073–3081. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Decaussin, G.; Sumner, S.; Ooka, T. N-terminal domain of BARF1 gene encoded by epstein-barr virus is essential for malignant transformation of rodent fibroblasts and activation of BCL-2. Oncogene 2001, 20, 1176–1185. [Google Scholar] [CrossRef] [Green Version]
- Karran, L.; Gao, Y.; Smith, P.R.; Griffin, B.E. Expression of a family of complementary-strand transcripts in epstein-barr virus-infected cells. Proc. Natl. Acad. Sci. USA 1992, 89, 8058–8062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- zur Hausen, A.; Brink, A.A.; Craanen, M.E.; Middeldorp, J.M.; Meijer, C.J.; van den Brule, A.J. Unique transcription pattern of epstein-barr virus (EBV) in EBV-carrying gastric adenocarcinomas: Expression of the transforming BARF1 gene. Cancer Res. 2000, 60, 2745–2748. [Google Scholar] [PubMed]
- Stevens, S.J.; Verkuijlen, S.A.; Hariwiyanto, B.; Harijadi; Paramita, D.K.; Fachiroh, J.; Adham, M.; Tan, I.B.; Haryana, S.M.; Middeldorp, J.M. Noninvasive diagnosis of nasopharyngeal carcinoma: Nasopharyngeal brushings reveal high epstein-barr virus DNA load and carcinoma-specific viral BARF1 mRNA. Int. J. Cancer 2006, 119, 608–614. [Google Scholar] [CrossRef]
- Rosemarie, Q.; Sugden, B. Epstein-barr virus: How its lytic phase contributes to oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Luftig, M.A. Dynamic epstein-barr virus gene expression on the path to B-cell transformation. Adv. Virus Res. 2014, 88, 279–313. [Google Scholar]
- Hu, L.; Lin, Z.; Wu, Y.; Dong, J.; Zhao, B.; Cheng, Y.; Huang, P.; Xu, L.; Xia, T.; Xiong, D.; et al. Comprehensive profiling of EBV gene expression in nasopharyngeal carcinoma through paired-end transcriptome sequencing. Front. Med. 2016, 10, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Matho, M.H.; Schlossman, A.; Meng, X.; Benhnia, M.R.; Kaever, T.; Buller, M.; Doronin, K.; Parker, S.; Peters, B.; Crotty, S.; et al. Structural and functional characterization of anti-A33 antibodies reveal a potent cross-species orthopoxviruses neutralizer. PLoS Pathog. 2015, 11, e1005148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Strong, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.; et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: Implications for possible immune adjuvant therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef]
- Feederle, R.; Kost, M.; Baumann, M.; Janz, A.; Drouet, E.; Hammerschmidt, W.; Delecluse, H.J. The epstein-barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000, 19, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Xue, S.A.; Labrecque, L.G.; Lu, Q.L.; Ong, S.K.; Lampert, I.A.; Kazembe, P.; Molyneux, E.; Broadhead, R.L.; Borgstein, E.; Griffin, B.E. Promiscuous expression of epstein-barr virus genes in burkitt’s lymphoma from the central African country Malawi. Int. J. Cancer 2002, 99, 635–643. [Google Scholar] [CrossRef]
- Cochet, C.; Martel-Renoir, D.; Grunewald, V.; Bosq, J.; Cochet, G.; Schwaab, G.; Bernaudin, J.F.; Joab, I. Expression of the epstein-barr virus immediate early gene, BZLF1, in nasopharyngeal carcinoma tumor cells. Virology 1993, 197, 358–365. [Google Scholar] [CrossRef]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of epstein-barr virus genomes and expression profiles in gastric adenocarcinoma. J. Virol. 2018, 92, e1003341. [Google Scholar] [CrossRef] [Green Version]
- Germini, D.; Sall, F.B.; Shmakova, A.; Wiels, J.; Dokudovskaya, S.; Drouet, E.; Vassetzky, Y. Oncogenic properties of the EBV ZEBRA protein. Cancers 2020, 12, 1479. [Google Scholar] [CrossRef]
- Marshall, W.L.; Yim, C.; Gustafson, E.; Graf, T.; Sage, D.R.; Hanify, K.; Williams, L.; Fingeroth, J.; Finberg, R.W. Epstein-barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with bax and bak. J. Virol. 1999, 73, 5181–5185. [Google Scholar] [CrossRef] [Green Version]
- Kelly, G.L.; Long, H.M.; Stylianou, J.; Thomas, W.A.; Leese, A.; Bell, A.I.; Bornkamm, G.W.; Mautner, J.; Rickinson, A.B.; Rowe, M. An epstein-barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: The Wp/BHRF1 link. PLoS Pathog. 2009, 5, e1000341. [Google Scholar] [CrossRef]
- Altmann, M.; Hammerschmidt, W. Epstein-barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beisser, P.S.; Verzijl, D.; Gruijthuijsen, Y.K.; Beuken, E.; Smit, M.J.; Leurs, R.; Bruggeman, C.A.; Vink, C. The epstein-barr virus BILF1 gene encodes a G protein-coupled receptor that inhibits phosphorylation of RNA-dependent protein kinase. J. Virol. 2005, 79, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Deng, W.; Hau, P.M.; Liu, J.; Lau, V.M.; Cheung, A.L.; Huen, M.S.; Tsao, S.W. Epstein-barr virus BZLF1 protein impairs accumulation of host DNA damage proteins at damage sites in response to DNA damage. Lab Investig. 2015, 95, 937–950. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zhu, J.; Xie, Z.; Liao, G.; Liu, J.; Chen, M.R.; Hu, S.; Woodard, C.; Lin, J.; Taverna, S.D.; et al. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe. 2011, 10, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.C.; Liu, M.T.; Chang, Y.T.; Fang, C.Y.; Chou, S.P.; Liao, H.W.; Kuo, K.L.; Hsu, S.L.; Chen, Y.R.; Wang, P.W.; et al. Epstein-barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010, 38, 1932–1949. [Google Scholar] [CrossRef]
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein-barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, O.F.; Pagano, J.S.; Whitehurst, C.B. The translesion polymerase pol eta is required for efficient epstein-barr virus infectivity and is regulated by the viral deubiquitinating enzyme BPLF1. J. Virol. 2017, 91, e00600-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, T.H.; Sitz, J.; Shen, Q.; Leblanc-Lacroix, A.; Campos, E.I.; Borozan, I.; Marcon, E.; Greenblatt, J.; Fradet-Turcotte, A.; Jin, D.Y.; et al. A screen for epstein-barr virus proteins that inhibit the DNA damage response reveals a novel histone binding protein. J. Virol. 2018, 92, e00262-18. [Google Scholar] [CrossRef] [Green Version]
- van Gent, M.; Braem, S.G.; de Jong, A.; Delagic, N.; Peeters, J.G.; Boer, I.G.; Moynagh, P.N.; Kremmer, E.; Wiertz, E.J.; Ovaa, H.; et al. Epstein-barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 2014, 10, e1003960. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Ressing, M.E.; van Leeuwen, D.; Pudney, V.A.; Horst, D.; Koppers-Lalic, D.; Croft, N.P.; Neefjes, J.J.; Rickinson, A.B.; Wiertz, E.J. A CD8+ T cell immune evasion protein specific to epstein-barr virus and its close relatives in old world primates. J. Exp. Med. 2007, 204, 1863–1873. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Currin, A.; Griffin, B.D.; Shannon-Lowe, C.; Thomas, W.A.; Ressing, M.E.; Wiertz, E.J.; Rowe, M. The epstein-barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog. 2009, 5, e1000255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, L.L.; Zuo, J.; Abbott, R.J.; Shannon-Lowe, C.; Tierney, R.J.; Hislop, A.D.; Rowe, M. Cooperation between epstein-barr virus immune evasion proteins spreads protection from CD8+ T cell recognition across all three phases of the lytic cycle. PLoS Pathog. 2014, 10, e1004322. [Google Scholar] [CrossRef]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.; Ressing, M.E. Host shutoff during productive epstein-barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Fossum, E.; Joo, C.H.; Inn, K.S.; Shin, Y.C.; Johannsen, E.; Hutt-Fletcher, L.M.; Hass, J.; Jung, J.U. Epstein-barr virus LF2: An antagonist to type I interferon. J. Virol. 2009, 83, 1140–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorley-Lawson, D.A.; Allday, M.J. The curious case of the tumour virus: 50 years of burkitt’s lymphoma. Nat. Rev. Microbiol. 2008, 6, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. Elife 2016, 5, e18270. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the world health organization classification of haematolymphoid tumours: Lymphoid neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Wu, T.C.; Mann, R.B.; Charache, P.; Hayward, S.D.; Staal, S.; Lambe, B.C.; Ambinder, R.F. Detection of EBV gene expression in reed-sternberg cells of hodgkin’s disease. Int. J. Cancer 1990, 46, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Coates, P.J.; Slavin, G.; D’Ardenne, A.J. Persistence of epstein-barr virus in reed-sternberg cells throughout the course of hodgkin’s disease. J. Pathol. 1991, 164, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.G.; Young, L.S. An etiological role for the epstein-barr virus in the pathogenesis of classical hodgkin lymphoma. Blood 2019, 134, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin disease: Hodgkin and reed-sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc. Natl. Acad. Sci. USA 1994, 91, 10962–10966. [Google Scholar] [CrossRef] [Green Version]
- Ok, C.Y.; Papathomas, T.G.; Medeiros, L.J.; Young, K.H. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood 2013, 122, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Malpica, L.; Marques-Piubelli, M.L.; Beltran, B.E.; Chavez, J.C.; Miranda, R.N.; Castillo, J.J. EBV-positive diffuse large B-cell lymphoma, not otherwise specified: 2022 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2022, 97, 951–965. [Google Scholar] [CrossRef]
- Kato, H.; Karube, K.; Yamamoto, K.; Takizawa, J.; Tsuzuki, S.; Yatabe, Y.; Kanda, T.; Katayama, M.; Ozawa, Y.; Ishitsuka, K.; et al. Gene expression profiling of epstein-barr virus-positive diffuse large B-cell lymphoma of the elderly reveals alterations of characteristic oncogenetic pathways. Cancer Sci. 2014, 105, 537–544. [Google Scholar] [CrossRef]
- Yoon, H.; Park, S.; Ju, H.; Ha, S.Y.; Sohn, I.; Jo, J.; Do, I.G.; Min, S.; Kim, S.J.; Kim, W.S.; et al. Integrated copy number and gene expression profiling analysis of epstein-barr virus-positive diffuse large B-cell lymphoma. Genes Chromosomes Cancer 2015, 54, 383–396. [Google Scholar] [CrossRef]
- White, R.E.; Ramer, P.C.; Naresh, K.N.; Meixlsperger, S.; Pinaud, L.; Rooney, C.; Savoldo, B.; Coutinho, R.; Bodor, C.; Gribben, J.; et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Invest. 2012, 122, 1487–1502. [Google Scholar] [CrossRef]
- Montes-Mojarro, I.A.; Fend, F.; Quintanilla-Martinez, L. EBV and the pathogenesis of NK/T cell lymphoma. Cancers 2021, 13, 1414. [Google Scholar] [CrossRef]
- Kanavaros, P.; Briere, J.; Emile, J.F.; Gaulard, P. Epstein-barr virus in T and natural killer (NK) cell non-hodgkin’s lymphomas. Leukemia 1996, 10 (Suppl. 2), s84–s87. [Google Scholar]
- Wang, H.; Fu, B.B.; Gale, R.P.; Liang, Y. NK-/T-cell lymphomas. Leukemia 2021, 35, 2460–2468. [Google Scholar] [CrossRef]
- Kingma, D.W.; Weiss, W.B.; Jaffe, E.S.; Kumar, S.; Frekko, K.; Raffeld, M. Epstein-barr virus latent membrane protein-1 oncogene deletions: Correlations with malignancy in epstein-barr virus—Associated lymphoproliferative disorders and malignant lymphomas. Blood 1996, 88, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, R.; Donahue, H.; Garcia, D.; Tan, J.; Shimizu, N.; Rice, A.P.; Ling, P.D. Epstein-barr virus BART9 miRNA modulates LMP1 levels and affects growth rate of nasal NK T cell lymphomas. PLoS ONE 2011, 6, e27271. [Google Scholar] [CrossRef]
- Huang, Y.; de Leval, L.; Gaulard, P. Molecular underpinning of extranodal NK/T-cell lymphoma. Best Pract. Res. Clin. Haematol. 2013, 26, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Bahri, R.; Boyer, F.; Halabi, M.A.; Chaunavel, A.; Feuillard, J.; Jaccard, A.; Ranger-Rogez, S. Epstein-barr virus (EBV) is mostly latent and clonal in angioimmunoblastic T cell lymphoma (AITL). Cancers 2022, 14, 2899. [Google Scholar] [CrossRef] [PubMed]
- Dunleavy, K.; Wilson, W.H. Angioimmunoblastic T-cell lymphoma: Immune modulation as a therapeutic strategy. Leuk Lymphoma 2007, 48, 449–451. [Google Scholar] [CrossRef]
- Bayda, N.; Tilloy, V.; Chaunavel, A.; Bahri, R.; Halabi, M.A.; Feuillard, J.; Jaccard, A.; Ranger-Rogez, S. Comprehensive epstein-barr virus transcriptome by RNA-sequencing in angioimmunoblastic T cell lymphoma (AITL) and other lymphomas. Cancers 2021, 13, 610. [Google Scholar] [CrossRef]
- Longnecker, R.M.; Kieff, E.; Cohen, J.I. Epstein-barr virus. In Fileds Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Wlliams, and Wilkins: Philadelphia, PA, USA, 2013; pp. 1898–1959. [Google Scholar]
- Rickinson, A.B.; Lo, K.W. Nasopharyngeal carcinoma: A history. In Nasopharyngeal Carcinoma; Lee, A.W.M., Lung, M.L., Ng, W.T., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 1–16. [Google Scholar]
- Pathmanathan, R.; Prasad, U.; Sadler, R.; Flynn, K.; Raab-Traub, N. Clonal proliferations of cells infected with epstein-barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N. Engl. J. Med. 1995, 333, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Lu, J.; Pei, Y.; Robertson, E.S. Transcriptome reprogramming of epstein-barr virus infected epithelial and B cells reveals distinct host-virus interaction profiles. Cell Death Dis. 2022, 13, 894. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Yip, Y.L.; Jia, L.; Deng, W.; Zheng, H.; Dai, W.; Ko, J.M.Y.; Lo, K.W.; Chung, G.T.Y.; Yip, K.Y.; et al. Establishment and characterization of new tumor xenografts and cancer cell lines from EBV-positive nasopharyngeal carcinoma. Nat. Commun. 2018, 9, 4663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.S.; Dawson, C.W.; Clark, D.; Rupani, H.; Busson, P.; Tursz, T.; Johnson, A.; Rickinson, A.B. Epstein-barr virus gene expression in nasopharyngeal carcinoma. J. Gen. Virol. 1988, 69 Pt 5, 1051–1065. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.; Yao, Q.Y.; Rickinson, A.B.; Young, L.S. Epstein-barr virus latent gene transcription in nasopharyngeal carcinoma cells: Coexpression of EBNA1, LMP1, and LMP2 transcripts. J. Virol. 1992, 66, 2689–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, S.; Dawson, C.W.; Takada, K.; Curnow, J.; Moody, C.A.; Sixbey, J.W.; Young, L.S. Epstein-barr virus-encoded LMP2A regulates viral and cellular gene expression by modulation of the NF-kappaB transcription factor pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 15730–15735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.C.; Meng, X.; Hazawa, M.; Nagata, Y.; Varela, A.M.; Xu, L.; Sato, Y.; Liu, L.Z.; Ding, L.W.; Sharma, A.; et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 2014, 46, 866–871. [Google Scholar] [CrossRef]
- Sun, K.; Jia, K.; Lv, H.; Wang, S.Q.; Wu, Y.; Lei, H.; Chen, X. EBV-positive gastric cancer: Current knowledge and future perspectives. Front. Oncol. 2020, 10, 583463. [Google Scholar] [CrossRef]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis shows that prevalence of epstein-barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Latency Pattern | Latency III | Latency II | Latency I | Latency 0 | |
|---|---|---|---|---|---|
| Genes | |||||
| EBNA1 | + | + | + | ND | |
| EBNA2 | + | ND | ND | ND | |
| EBNA3s | + | ND | ND | ND | |
| EBNA-LP | + | ND | ND | ND | |
| LMP1 | + | + | ND | ND | |
| LMP2 | + | + | ND | ND | |
| EBERs | + | + | + | + | |
| BHRF1 miRNAs | + | ND | ND | ND | |
| BARTs miRNAs | + | + | + | + | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Robertson, E.S. Epstein–Barr Virus History and Pathogenesis. Viruses 2023, 15, 714. https://doi.org/10.3390/v15030714
Yu H, Robertson ES. Epstein–Barr Virus History and Pathogenesis. Viruses. 2023; 15(3):714. https://doi.org/10.3390/v15030714
Chicago/Turabian StyleYu, Hui, and Erle S. Robertson. 2023. "Epstein–Barr Virus History and Pathogenesis" Viruses 15, no. 3: 714. https://doi.org/10.3390/v15030714
APA StyleYu, H., & Robertson, E. S. (2023). Epstein–Barr Virus History and Pathogenesis. Viruses, 15(3), 714. https://doi.org/10.3390/v15030714