Hepatitis C Virus-Induced Mitochondrial Dysfunctions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
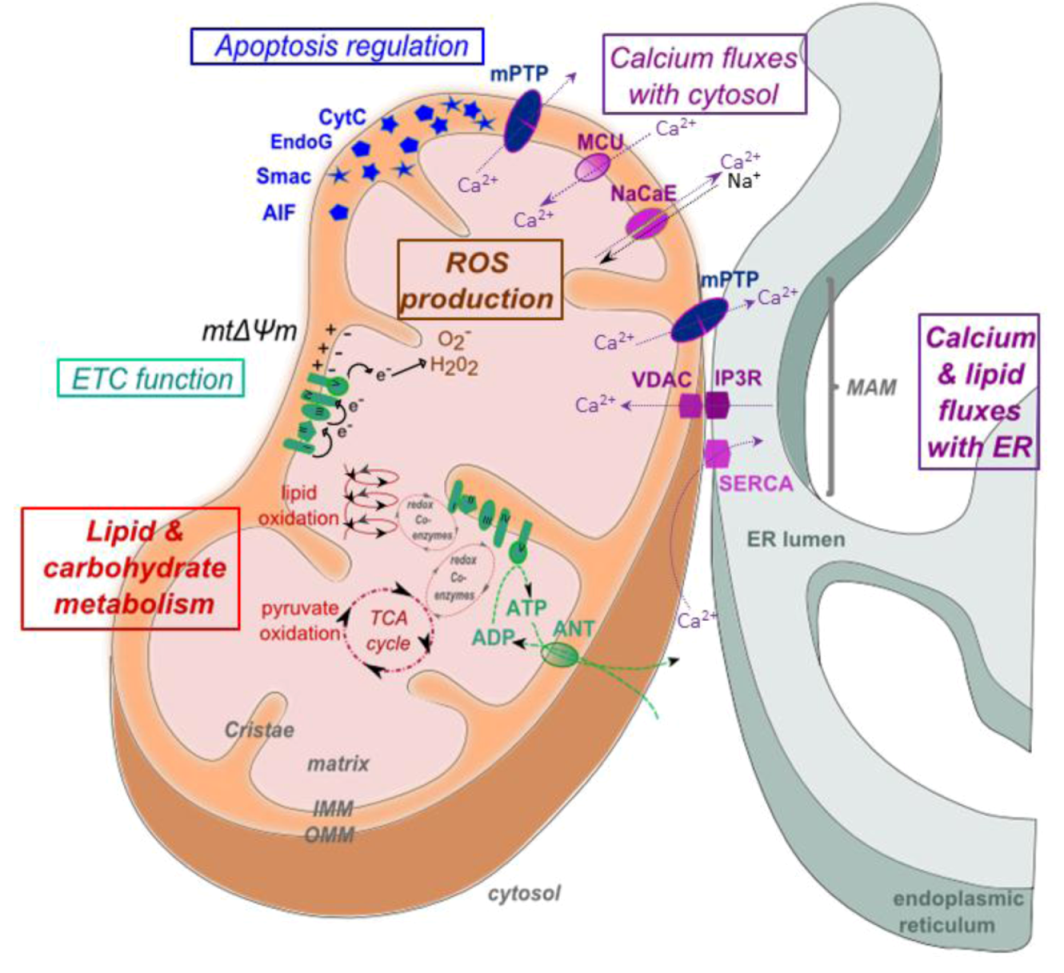
2. Mitochondrial Structure and Function

3. Mitochondrial Morphology in Productive HCV Infection
4. Physical Interactions between HCV and Mitochondria
5. Mitochondrial Functions Altered by HCV Infection
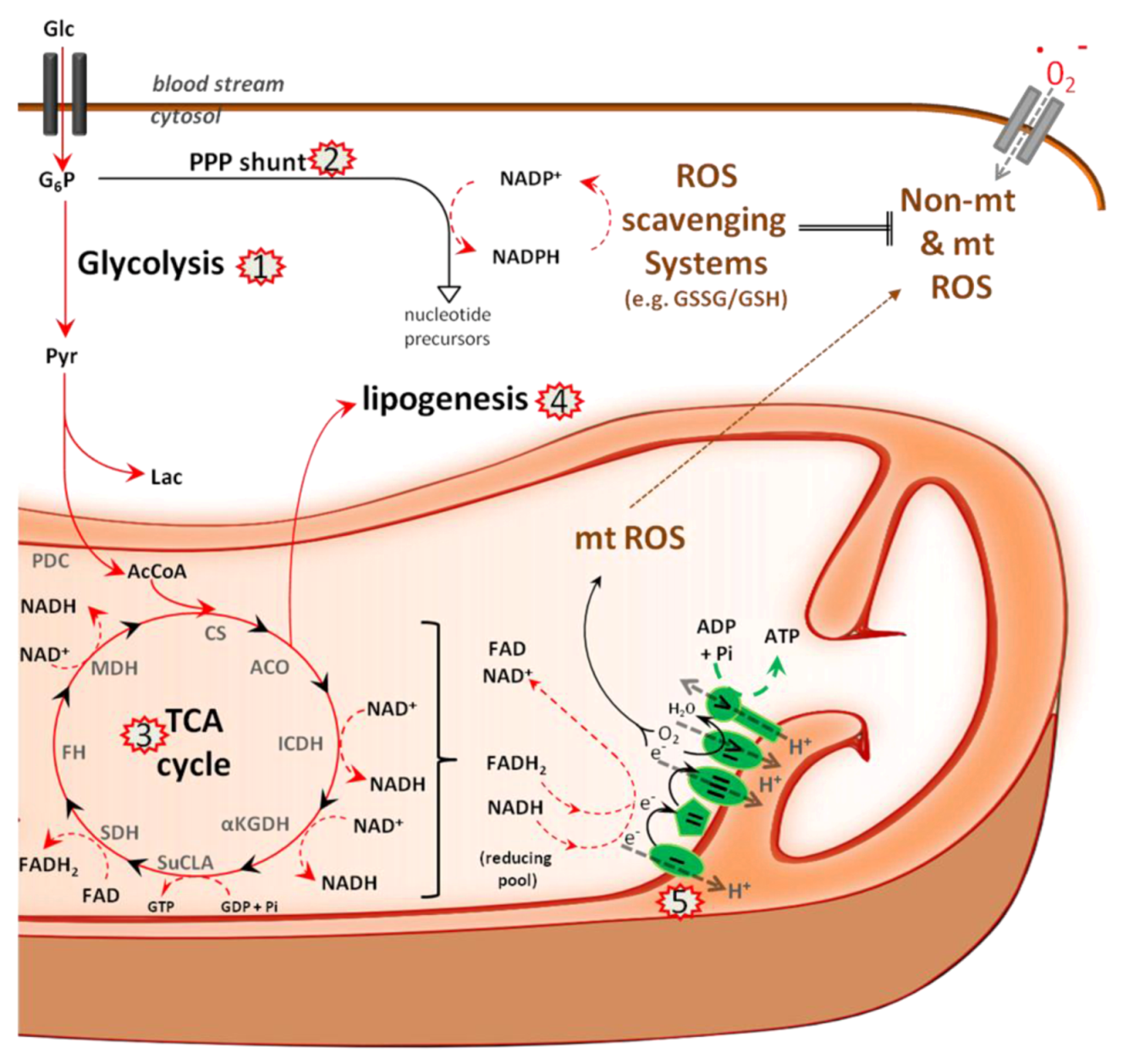
5.1. Metabolism


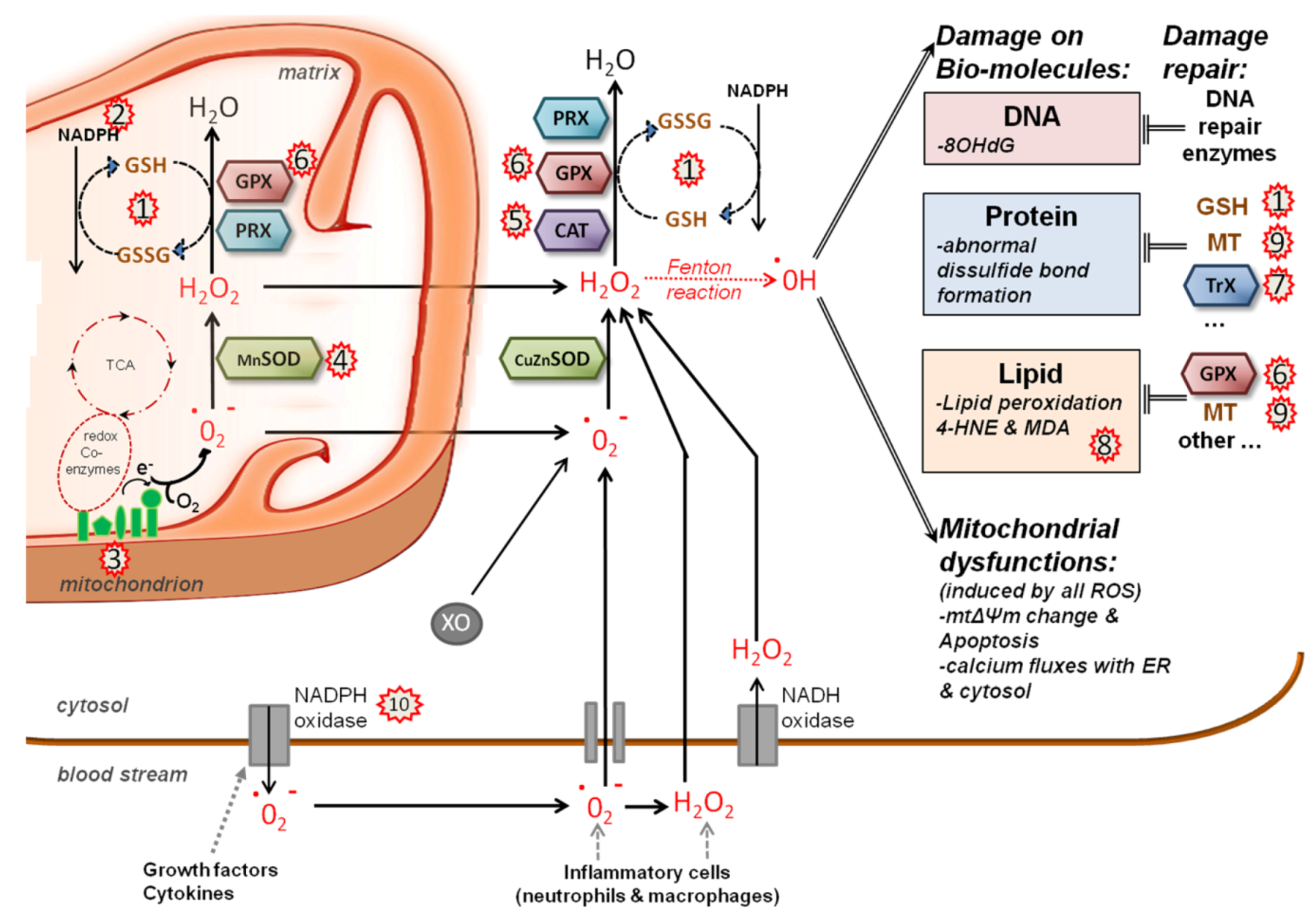
5.2. Redox System
5.3. Calcium Signaling

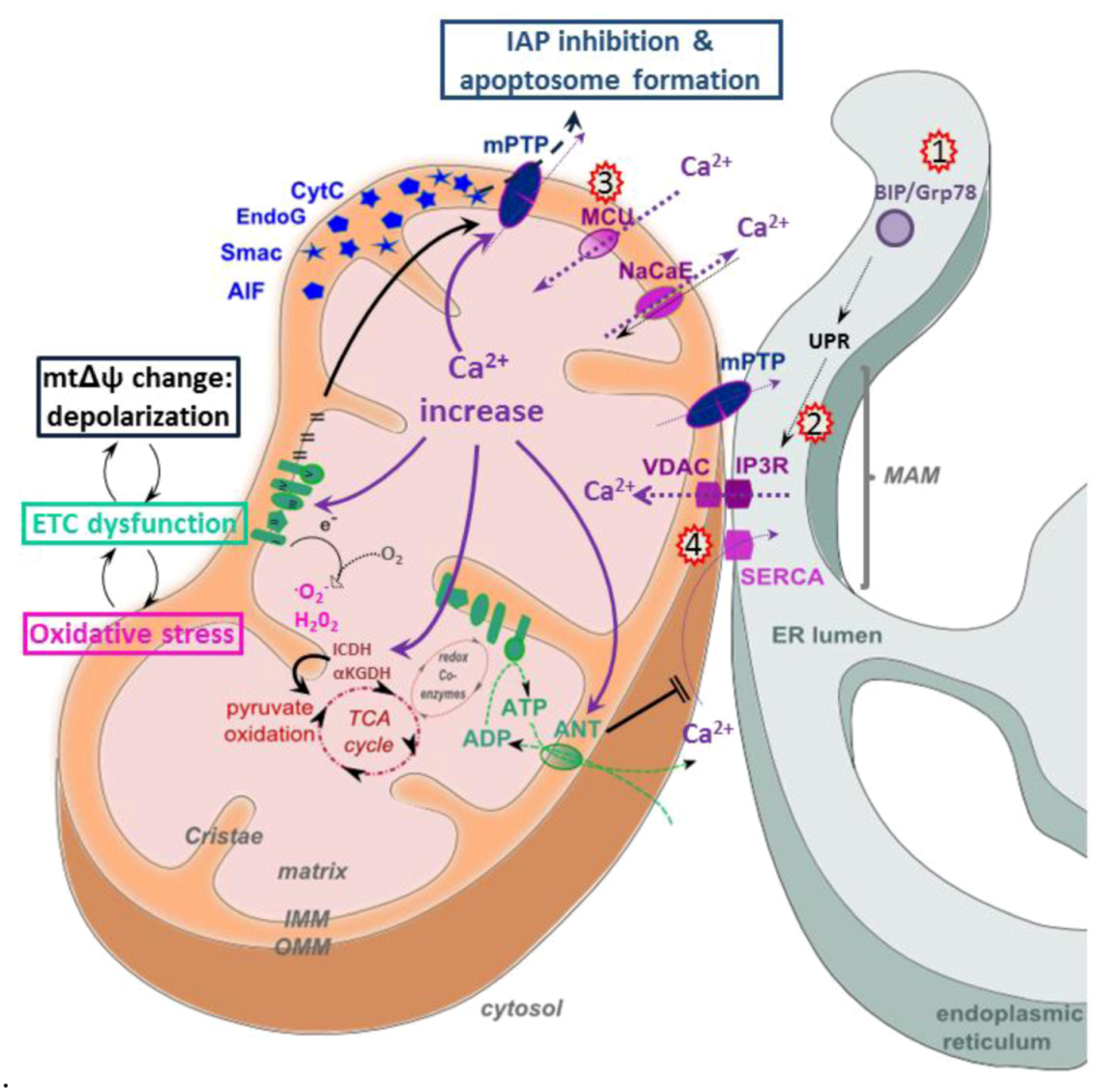
5.4. Apoptosis

6. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Lemon, S.M.; Walker, C.M.; Alter, M.J.; Yi, M.–K. Hepatitis c virus. In Fields Virology, 5th; Knipe, D.M., Howley, P.M., Eds.; 2007; pp. 1253–1304. [Google Scholar]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in il28b predicts hepatitis c treatment–induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar]
- Alaei, M.; Negro, F. Hepatitis c virus and glucose and lipid metabolism. Diabetes Metab. 2008, 34, 692–700. [Google Scholar] [CrossRef]
- Bartosch, B. Hepatitis c virus and its complex interplay with hepatic glucose and lipid metabolism. J. Hepatol. 2009, 50, 845–847. [Google Scholar] [CrossRef]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis c virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Bartosch, B. Hepatitis b and c viruses and hepatocellular carcinoma. Viruses 2010, 2, 1504–1509. [Google Scholar] [CrossRef]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis c virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis c virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef]
- Goldenthal, M.J.; Marin–Garcia, J. Mitochondrial signaling pathways: A receiver/integrator organelle. Mol. Cell Biochem. 2004, 262, 1–16. [Google Scholar] [CrossRef]
- Duchen, M.R. Roles of mitochondria in health and disease. Diabetes 2004, 53, 96–102. [Google Scholar] [CrossRef]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria–associated membrane (mam). Biochim. Biophys. Acta. 2013, 1833, 213–224. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone–mediated coupling of endoplasmic reticulum and mitochondrial ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Filippin, L.; Magalhaes, P.J.; Di Benedetto, G.; Colella, M.; Pozzan, T. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J. Biol. Chem. 2003, 278, 39224–39234. [Google Scholar]
- Rusinol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria–associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre–golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar]
- Rimessi, A.; Giorgi, C.; Pinton, P.; Rizzuto, R. The versatility of mitochondrial calcium signals: From stimulation of cell metabolism to induction of cell death. Biochim. Biophys. Acta. 1777, 808–816. [Google Scholar]
- Balaban, R.S. Cardiac energy metabolism homeostasis: Role of cytosolic calcium. J. Mol. Cell Cardiol. 2002, 34, 1259–1271. [Google Scholar] [CrossRef]
- Denton, R.M.; McCormack, J.G. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu. Rev. Physiol. 1990, 52, 451–466. [Google Scholar] [CrossRef]
- Liu, T.; O'Rourke, B. Regulation of mitochondrial ca2+ and its effects on energetics and redox balance in normal and failing heart. J. Bioenerg. Biomembr. 2009, 41, 127–132. [Google Scholar] [CrossRef]
- Galley, H.F. Bench–to–bedside review: Targeting antioxidants to mitochondria in sepsis. Crit. Care. 2010, 14, 230. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ros generation and its regulation: Mechanisms involved in h(2)o(2) signaling. Antioxid Redox Signal 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Hansford, R.G.; Hogue, B.A.; Mildaziene, V. Dependence of h2o2 formation by rat heart mitochondria on substrate availability and donor age. J. Bioenerg. Biomembr. 1997, 29, 89–95. [Google Scholar] [CrossRef]
- Tahara, E.B.; Navarete, F.D.; Kowaltowski, A.J. Tissue–, substrate–, and site–specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol. Med. 2009, 46, 1283–1297. [Google Scholar] [CrossRef]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial ca2+ and ros. Front Biosci. 2009, 14, 1197–1218. [Google Scholar]
- Karbowski, M.; Youle, R.J. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003, 10, 870–880. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.Y.; Lee, S.; Kim, S.W.; Sohn, S.; Kim, K.; Cho, H. Hepatitis b virus x protein induces perinuclear mitochondrial clustering in microtubule– and dynein–dependent manners. J. Virol. 2007, 81, 1714–1726. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chiang, S.F.; Lin, T.Y.; Chiou, S.H.; Chow, K.C. Hiv–1 vpr triggers mitochondrial destruction by impairing mfn2–mediated er–mitochondria interaction. PLoS One 2012, 7, e33657. [Google Scholar]
- Radovanovic, J.; Todorovic, V.; Boricic, I.; Jankovic–Hladni, M.; Korac, A. Comparative ultrastructural studies on mitochondrial pathology in the liver of aids patients: Clusters of mitochondria, protuberances, "minimitochondria," vacuoles, and virus–like particles. Ultrastruct. Pathol. 1999, 23, 19–24. [Google Scholar] [CrossRef]
- D'Agostino, D.M.; Ranzato, L.; Arrigoni, G.; Cavallari, I.; Belleudi, F.; Torrisi, M.R.; Silic–Benussi, M.; Ferro, T.; Petronilli, V.; Marin, O.; et al. Mitochondrial alterations induced by the p13ii protein of human t–cell leukemia virus type 1. Critical role of arginine residues. J. Biol. Chem. 2002, 277, 34424–34433. [Google Scholar] [CrossRef]
- Yamada, H.; Chounan, R.; Higashi, Y.; Kurihara, N.; Kido, H. Mitochondrial targeting sequence of the influenza a virus pb1–f2 protein and its function in mitochondria. FEBS Lett. 2004, 578, 331–336. [Google Scholar] [CrossRef]
- Boya, P.; Pauleau, A.L.; Poncet, D.; Gonzalez–Polo, R.A.; Zamzami, N.; Kroemer, G. Viral proteins targeting mitochondria: Controlling cell death. Biochim. Biophys. Acta. 2004, 1659, 178–189. [Google Scholar] [CrossRef]
- Li, Y.; Boehning, D.F.; Qian, T.; Popov, V.L.; Weinman, S.A. Hepatitis c virus core protein increases mitochondrial ros production by stimulation of ca2+ uniporter activity. FASEB J. 2007, 21, 2474–2485. [Google Scholar] [CrossRef]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis c virus–associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog 2010, 6, e1000719. [Google Scholar] [CrossRef]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis c virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef]
- Joyce, M.A.; Walters, K.A.; Lamb, S.E.; Yeh, M.M.; Zhu, L.F.; Kneteman, N.; Doyle, J.S.; Katze, M.G.; Tyrrell, D.L. Hcv induces oxidative and er stress, and sensitizes infected cells to apoptosis in scid/alb–upa mice. PLoS Pathog 2009, 5, e1000291. [Google Scholar] [CrossRef]
- Merquiol, E.; Uzi, D.; Mueller, T.; Goldenberg, D.; Nahmias, Y.; Xavier, R.J.; Tirosh, B.; Shibolet, O. Hcv causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One 2011, 6, e24660. [Google Scholar]
- Tardif, K.D.; Waris, G.; Siddiqui, A. Hepatitis c virus, er stress, and oxidative stress. Trends Microbiol. 2005, 13, 159–163. [Google Scholar] [CrossRef]
- Piccoli, C.; Scrima, R.; Quarato, G.; D'Aprile, A.; Ripoli, M.; Lecce, L.; Boffoli, D.; Moradpour, D.; Capitanio, N. Hepatitis c virus protein expression causes calcium–mediated mitochondrial bioenergetic dysfunction and nitro–oxidative stress. Hepatology 2007, 46, 58–65. [Google Scholar] [CrossRef]
- Wang, T.; Weinman, S.A. Causes and consequences of mitochondrial reactive oxygen species generation in hepatitis c. J. Gastroenterol. Hepatol. 2006, 21 Suppl 3, S34–S37. [Google Scholar] [CrossRef]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis c virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ros) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar]
- Ait–Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis c virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar] [CrossRef]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis c virus rna on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar]
- Quan, W.; Jung, H.S.; Lee, M.S. Role of autophagy in the progression from obesity to diabetes and in the control of energy balance. Arch. Pharm. Res. 2013. [Google Scholar]
- Barbaro, G.; Di Lorenzo, G.; Asti, A.; Ribersani, M.; Belloni, G.; Grisorio, B.; Filice, G.; Barbarini, G. Hepatocellular mitochondrial alterations in patients with chronic hepatitis c: Ultrastructural and biochemical findings. Am. J. Gastroenterol. 1999, 94, 2198–2205. [Google Scholar] [CrossRef]
- Deng, L.; Adachi, T.; Kitayama, K.; Bungyoku, Y.; Kitazawa, S.; Ishido, S.; Shoji, I.; Hotta, H. Hepatitis c virus infection induces apoptosis through a bax–triggered, mitochondrion–mediated, caspase 3–dependent pathway. J. Virol. 2008, 82, 10375–10385. [Google Scholar]
- Chu, V.C.; Bhattacharya, S.; Nomoto, A.; Lin, J.; Zaidi, S.K.; Oberley, T.D.; Weinman, S.A.; Azhar, S.; Huang, T.T. Persistent expression of hepatitis c virus non–structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS One 2011, 6, e28551. [Google Scholar]
- Nomura–Takigawa, Y.; Nagano–Fujii, M.; Deng, L.; Kitazawa, S.; Ishido, S.; Sada, K.; Hotta, H. Non–structural protein 4a of hepatitis c virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria–mediated apoptosis. J. Gen. Virol. 2006, 87, 1935–1945. [Google Scholar] [CrossRef]
- Barba, G.; Harper, F.; Harada, T.; Kohara, M.; Goulinet, S.; Matsuura, Y.; Eder, G.; Schaff, Z.; Chapman, M.J.; Miyamura, T.; et al. Hepatitis c virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 1200–1205. [Google Scholar] [CrossRef]
- Okuda, M.; Li, K.; Beard, M.R.; Showalter, L.A.; Scholle, F.; Lemon, S.M.; Weinman, S.A. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis c virus core protein. Gastroenterology 2002, 122, 366–375. [Google Scholar] [CrossRef]
- Suzuki, R.; Sakamoto, S.; Tsutsumi, T.; Rikimaru, A.; Tanaka, K.; Shimoike, T.; Moriishi, K.; Iwasaki, T.; Mizumoto, K.; Matsuura, Y.; et al. Molecular determinants for subcellular localization of hepatitis c virus core protein. J. Virol. 2005, 79, 1271–1281. [Google Scholar] [CrossRef]
- Schwer, B.; Ren, S.; Pietschmann, T.; Kartenbeck, J.; Kaehlcke, K.; Bartenschlager, R.; Yen, T.S.; Ott, M. Targeting of hepatitis c virus core protein to mitochondria through a novel c–terminal localization motif. J. Virol. 2004, 78, 7958–7968. [Google Scholar] [CrossRef]
- Kasprzak, A.; Seidel, J.; Biczysko, W.; Wysocki, J.; Spachacz, R.; Zabel, M. Intracellular localization of ns3 and c proteins in chronic hepatitis c. Liver Int. 2005, 25, 896–903. [Google Scholar] [CrossRef]
- Rouille, Y.; Helle, F.; Delgrange, D.; Roingeard, P.; Voisset, C.; Blanchard, E.; Belouzard, S.; McKeating, J.; Patel, A.H.; Maertens, G.; et al. Subcellular localization of hepatitis c virus structural proteins in a cell culture system that efficiently replicates the virus. J. Virol. 2006, 80, 2832–2841. [Google Scholar] [CrossRef]
- Lai, C.K.; Jeng, K.S.; Machida, K.; Lai, M.M. Hepatitis c virus egress and release depend on endosomal trafficking of core protein. J. Virol. 2010, 84, 11590–11598. [Google Scholar] [CrossRef]
- Wang, T.; Campbell, R.V.; Yi, M.K.; Lemon, S.M.; Weinman, S.A. Role of hepatitis c virus core protein in viral–induced mitochondrial dysfunction. J. Viral Hepat. 2010, 17, 784–793. [Google Scholar] [CrossRef]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial–associated endoplasmic reticulum membranes (mam) form innate immune synapses and are targeted by hepatitis c virus. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 14590–14595. [Google Scholar]
- Mottola, G.; Cardinali, G.; Ceccacci, A.; Trozzi, C.; Bartholomew, L.; Torrisi, M.R.; Pedrazzini, E.; Bonatti, S.; Migliaccio, G. Hepatitis c virus nonstructural proteins are localized in a modified endoplasmic reticulum of cells expressing viral subgenomic replicons. Virology 2002, 293, 31–43. [Google Scholar] [CrossRef]
- Wolk, B.; Sansonno, D.; Krausslich, H.G.; Dammacco, F.; Rice, C.M.; Blum, H.E.; Moradpour, D. Subcellular localization, stability, and trans–cleavage competence of the hepatitis c virus ns3–ns4a complex expressed in tetracycline–regulated cell lines. J. Virol. 2000, 74, 2293–2304. [Google Scholar] [CrossRef]
- Griffin, S.; Clarke, D.; McCormick, C.; Rowlands, D.; Harris, M. Signal peptide cleavage and internal targeting signals direct the hepatitis c virus p7 protein to distinct intracellular membranes. J. Virol. 2005, 79, 15525–15536. [Google Scholar] [CrossRef]
- Abdalla, M.Y.; Ahmad, I.M.; Spitz, D.R.; Schmidt, W.N.; Britigan, B.E. Hepatitis c virus–core and non structural proteins lead to different effects on cellular antioxidant defenses. J. Med. Virol. 2005, 76, 489–497. [Google Scholar] [CrossRef]
- Fujinaga, H.; Tsutsumi, T.; Yotsuyanagi, H.; Moriya, K.; Koike, K. Hepatocarcinogenesis in hepatitis c: Hcv shrewdly exacerbates oxidative stress by modulating both production and scavenging of reactive oxygen species. Oncology 2011, 81 Suppl 1, 11–17. [Google Scholar] [CrossRef]
- Morbitzer, M.; Herget, T. Expression of gastrointestinal glutathione peroxidase is inversely correlated to the presence of hepatitis c virus subgenomic rna in human liver cells. J. Biol. Chem. 2005, 280, 8831–8841. [Google Scholar] [CrossRef]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis c virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and stat3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef]
- Li, K.; Prow, T.; Lemon, S.M.; Beard, M.R. Cellular response to conditional expression of hepatitis c virus core protein in huh7 cultured human hepatoma cells. Hepatology 2002, 35, 1237–1246. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Emerson, S.U.; Korzeniowska, A.; Jendrysik, M.A.; Leto, T.L. Hepatitis c virus (hcv) proteins induce nadph oxidase 4 expression in a transforming growth factor beta–dependent manner: A new contributor to hcv–induced oxidative stress. J. Virol. 2009, 83, 12934–12946. [Google Scholar] [CrossRef]
- de Mochel, N.S.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte nad(p)h oxidases as an endogenous source of reactive oxygen species during hepatitis c virus infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef]
- Stuyver, L.J.; Whitaker, T.; McBrayer, T.R.; Hernandez–Santiago, B.I.; Lostia, S.; Tharnish, P.M.; Ramesh, M.; Chu, C.K.; Jordan, R.; Shi, J.; et al. Ribonucleoside analogue that blocks replication of bovine viral diarrhea and hepatitis c viruses in culture. Antimicrob. Agents Chemother. 2003, 47, 244–254. [Google Scholar] [CrossRef]
- Ando, T.; Imamura, H.; Suzuki, R.; Aizaki, H.; Watanabe, T.; Wakita, T.; Suzuki, T. Visualization and measurement of atp levels in living cells replicating hepatitis c virus genome rna. PLoS Pathog 2012, 8, e1002561. [Google Scholar] [CrossRef]
- Rasmussen, A.L.; Diamond, D.L.; McDermott, J.E.; Gao, X.; Metz, T.O.; Matzke, M.M.; Carter, V.S.; Belisle, S.E.; Korth, M.J.; Waters, K.M.; et al. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme a delta isomerase, required for hepatitis c virus replication and likely pathogenesis. J. Virol. 2011, 85, 11646–11654. [Google Scholar] [CrossRef]
- Dharancy, S.; Malapel, M.; Perlemuter, G.; Roskams, T.; Cheng, Y.; Dubuquoy, L.; Podevin, P.; Conti, F.; Canva, V.; Philippe, D.; et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis c virus infection. Gastroenterology 2005, 128, 334–342. [Google Scholar] [CrossRef]
- Cheng, Y.; Dharancy, S.; Malapel, M.; Desreumaux, P. Hepatitis c virus infection down–regulates the expression of peroxisome proliferator–activated receptor alpha and carnitine palmitoyl acyl–coa transferase 1a. World J. Gastroenterol 2005, 11, 7591–7596. [Google Scholar]
- Syed, G.H.; Siddiqui, A. Effects of hypolipidemic agent nordihydroguaiaretic acid on lipid droplets and hepatitis c virus. Hepatology 2011, 54, 1936–1946. [Google Scholar] [CrossRef]
- Shimoda, R.; Nagashima, M.; Sakamoto, M.; Yamaguchi, N.; Hirohashi, S.; Yokota, J.; Kasai, H. Increased formation of oxidative DNA damage, 8–hydroxydeoxyguanosine, in human livers with chronic hepatitis. Cancer Res. 1994, 54, 3171–3172. [Google Scholar]
- Mahmood, S.; Kawanaka, M.; Kamei, A.; Izumi, A.; Nakata, K.; Niiyama, G.; Ikeda, H.; Hanano, S.; Suehiro, M.; Togawa, K.; et al. Immunohistochemical evaluation of oxidative stress markers in chronic hepatitis c. Antioxid. Redox Signal 2004, 6, 19–24. [Google Scholar] [CrossRef]
- Fujita, N.; Sugimoto, R.; Ma, N.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Kawanishi, S.; Watanabe, S.; Kaito, M.; Takei, Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis b and c. J. Viral Hepat. 2008, 15, 498–507. [Google Scholar] [CrossRef]
- Farinati, F.; Cardin, R.; De Maria, N.; Della Libera, G.; Marafin, C.; Lecis, E.; Burra, P.; Floreani, A.; Cecchetto, A.; Naccarato, R. Iron storage, lipid peroxidation and glutathione turnover in chronic anti–hcv positive hepatitis. J. Hepatol. 1995, 22, 449–456. [Google Scholar] [CrossRef]
- Kageyama, F.; Kobayashi, Y.; Kawasaki, T.; Toyokuni, S.; Uchida, K.; Nakamura, H. Successful interferon therapy reverses enhanced hepatic iron accumulation and lipid peroxidation in chronic hepatitis c. Am. J. Gastroenterol. 2000, 95, 1041–1050. [Google Scholar] [CrossRef]
- Paradis, V.; Mathurin, P.; Kollinger, M.; Imbert–Bismut, F.; Charlotte, F.; Piton, A.; Opolon, P.; Holstege, A.; Poynard, T.; Bedossa, P. In situ detection of lipid peroxidation in chronic hepatitis c: Correlation with pathological features. J Clin Pathol 1997, 50, 401–406. [Google Scholar]
- De Maria, N.; Colantoni, A.; Fagiuoli, S.; Liu, G.J.; Rogers, B.K.; Farinati, F.; Van Thiel, D.H.; Floyd, R.A. Association between reactive oxygen species and disease activity in chronic hepatitis c. Free Radic. Biol. Med. 1996, 21, 291–295. [Google Scholar] [CrossRef]
- Romero, F.J.; Bosch–Morell, F.; Romero, M.J.; Jareno, E.J.; Romero, B.; Marin, N.; Roma, J. Lipid peroxidation products and antioxidants in human disease. Environ. Health Perspect. 1998, 106 Suppl 5, 1229–1234. [Google Scholar]
- Yadav, D.; Hertan, H.I.; Schweitzer, P.; Norkus, E.P.; Pitchumoni, C.S. Serum and liver micronutrient antioxidants and serum oxidative stress in patients with chronic hepatitis c. Am. J. Gastroenterol. 2002, 97, 2634–2639. [Google Scholar] [CrossRef]
- Ko, W.S.; Guo, C.H.; Yeh, M.S.; Lin, L.Y.; Hsu, G.S.; Chen, P.C.; Luo, M.C.; Lin, C.Y. Blood micronutrient, oxidative stress, and viral load in patients with chronic hepatitis c. World J. Gastroenterol. 2005, 11, 4697–4702. [Google Scholar]
- Nakashima, T.; Sumida, Y.; Yoh, T.; Kakisaka, Y.; Nakajima, Y.; Ishikawa, H.; Mitsuyoshi, H.; Kashima, K.; Nakamura, H.; Yodoi, J. Thioredoxin levels in the sera of untreated viral hepatitis patients and those treated with glycyrrhizin or ursodeoxycholic acid. Antioxid Redox Signal 2000, 2, 687–694. [Google Scholar] [CrossRef]
- Thorburn, D.; Curry, G.; Spooner, R.; Spence, E.; Oien, K.; Halls, D.; Fox, R.; McCruden, E.A.; MacSween, R.N.; Mills, P.R. The role of iron and haemochromatosis gene mutations in the progression of liver disease in chronic hepatitis c. Gut 2002, 50, 248–252. [Google Scholar] [CrossRef]
- Izumi, N.; Enomoto, N.; Uchihara, M.; Murakami, T.; Ono, K.; Noguchi, O.; Miyake, S.; Nouchi, T.; Fujisawa, K.; Marumo, F.; et al. Hepatic iron contents and response to interferon-alpha in patients with chronic hepatitis c. Relationship to genotypes of hepatitis c virus. Dig. Dis. Sci. 1996, 41, 989–994. [Google Scholar]
- Hezode, C.; Cazeneuve, C.; Coue, O.; Roudot–Thoraval, F.; Lonjon, I.; Bastie, A.; Duvoux, C.; Pawlotsky, J.M.; Zafrani, E.S.; Amselem, S.; et al. Liver iron accumulation in patients with chronic active hepatitis c: Prevalence and role of hemochromatosis gene mutations and relationship with hepatic histological lesions. J. Hepatol. 1999, 31, 979–984. [Google Scholar] [CrossRef]
- Vendemiale, G.; Grattagliano, I.; Portincasa, P.; Serviddio, G.; Palasciamo, G.; Altomare, E. Oxidative stress in symptom–free hcv carriers: Relation with alt flare–up. Eur. J. Clin. Invest. 2001, 31, 54–63. [Google Scholar]
- Diamond, D.L.; Jacobs, J.M.; Paeper, B.; Proll, S.C.; Gritsenko, M.A.; Carithers, R.L., Jr.; Larson, A.M.; Yeh, M.M.; Camp, D.G., 2nd; Smith, R.D.; et al. Proteomic profiling of human liver biopsies: Hepatitis c virus–induced fibrosis and mitochondrial dysfunction. Hepatology 2007, 46, 649–657. [Google Scholar] [CrossRef]
- Chan, S.W.; Egan, P.A. Hepatitis c virus envelope proteins regulate chop via induction of the unfolded protein response. FASEB J. 2005, 19, 1510–1512. [Google Scholar]
- Qadri, I.; Iwahashi, M.; Capasso, J.M.; Hopken, M.W.; Flores, S.; Schaack, J.; Simon, F.R. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis c virus replication: Role of jnk, p38 mapk and ap–1. Biochem J 2004, 378, 919–928. [Google Scholar]
- Moriya, K.; Nakagawa, K.; Santa, T.; Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Miyazawa, T.; Ishibashi, K.; Horie, T.; et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis c virus–associated hepatocarcinogenesis. Cancer Res. 2001, 61, 4365–4370. [Google Scholar]
- Lin, W.; Tsai, W.L.; Shao, R.X.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J.Y.; et al. Hepatitis c virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappab–dependent manner. Gastroenterology 2010, 138, 2509–2518, 2518 e2501.. [Google Scholar] [CrossRef]
- Polyak, S.J.; Morishima, C.; Lohmann, V.; Pal, S.; Lee, D.Y.; Liu, Y.; Graf, T.N.; Oberlies, N.H. Identification of hepatoprotective flavonolignans from silymarin. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 5995–5999. [Google Scholar]
- Dionisio, N.; Garcia–Mediavilla, M.V.; Sanchez–Campos, S.; Majano, P.L.; Benedicto, I.; Rosado, J.A.; Salido, G.M.; Gonzalez–Gallego, J. Hepatitis c virus ns5a and core proteins induce oxidative stress–mediated calcium signalling alterations in hepatocytes. J. Hepatol. 2009, 50, 872–882. [Google Scholar] [CrossRef]
- Korenaga, M.; Okuda, M.; Otani, K.; Wang, T.; Li, Y.; Weinman, S.A. Mitochondrial dysfunction in hepatitis c. J. Clin. Gastroenterol. 2005, 39, S162–S166. [Google Scholar] [CrossRef]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis c virus ns5a protein alters intracellular calcium levels, induces oxidative stress, and activates stat–3 and nf–kappa b. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 9599–9604. [Google Scholar] [CrossRef]
- Hara, Y.; Hino, K.; Okuda, M.; Furutani, T.; Hidaka, I.; Yamaguchi, Y.; Korenaga, M.; Li, K.; Weinman, S.A.; Lemon, S.M.; et al. Hepatitis c virus core protein inhibits deoxycholic acid–mediated apoptosis despite generating mitochondrial reactive oxygen species. J. Gastroenterol. 2006, 41, 257–268. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis c virus proteins activate nrf2/are pathway by distinct ros–dependent and independent mechanisms in huh7 cells. PLoS One 2011, 6, e24957. [Google Scholar]
- Machida, K.; Cheng, K.T.; Sung, V.M.; Lee, K.J.; Levine, A.M.; Lai, M.M. Hepatitis c virus infection activates the immunologic (type ii) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J. Virol. 2004, 78, 8835–8843. [Google Scholar]
- Ghaziani, T.; Shan, Y.; Lambrecht, R.W.; Donohue, S.E.; Pietschmann, T.; Bartenschlager, R.; Bonkovsky, H.L. Hcv proteins increase expression of heme oxygenase–1 (ho–1) and decrease expression of bach1 in human hepatoma cells. J. Hepatol. 2006, 45, 5–12. [Google Scholar] [CrossRef]
- Abdalla, M.Y.; Britigan, B.E.; Wen, F.; Icardi, M.; McCormick, M.L.; LaBrecque, D.R.; Voigt, M.; Brown, K.E.; Schmidt, W.N. Down–regulation of heme oxygenase–1 by hepatitis c virus infection in vivo and by the in vitro expression of hepatitis c core protein. J. Infect. Dis. 2004, 190, 1109–1118. [Google Scholar] [CrossRef]
- Garcia–Mediavilla, M.V.; Sanchez–Campos, S.; Gonzalez–Perez, P.; Gomez–Gonzalo, M.; Majano, P.L.; Lopez–Cabrera, M.; Clemente, G.; Garcia–Monzon, C.; Gonzalez–Gallego, J. Differential contribution of hepatitis c virus ns5a and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte–derived cells. J. Hepatol. 2005, 43, 606–613. [Google Scholar] [CrossRef]
- Robinson, L.C.; Marchant, J.S. Enhanced ca2+ leak from er ca2+ stores induced by hepatitis c ns5a protein. Biochem. Biophys. Res. Commun. 2008, 368, 593–599. [Google Scholar] [CrossRef]
- Roe, B.; Kensicki, E.; Mohney, R.; Hall, W.W. Metabolomic profile of hepatitis c virus–infected hepatocytes. PLoS One 2011, 6, e23641. [Google Scholar]
- Yano, M.; Ikeda, M.; Abe, K.; Dansako, H.; Ohkoshi, S.; Aoyagi, Y.; Kato, N. Comprehensive analysis of the effects of ordinary nutrients on hepatitis c virus rna replication in cell culture. Antimicrob. Agents Chemother. 2007, 51, 2016–2027. [Google Scholar] [CrossRef]
- Nakamura, M.; Saito, H.; Ikeda, M.; Hokari, R.; Kato, N.; Hibi, T.; Miura, S. An antioxidant resveratrol significantly enhanced replication of hepatitis c virus. World J. Gastroenterol. 2010, 16, 184–192. [Google Scholar] [CrossRef]
- Kuroki, M.; Ariumi, Y.; Ikeda, M.; Dansako, H.; Wakita, T.; Kato, N. Arsenic trioxide inhibits hepatitis c virus rna replication through modulation of the glutathione redox system and oxidative stress. J. Virol. 2009, 83, 2338–2348. [Google Scholar] [CrossRef]
- Huang, H.; Chen, Y.; Ye, J. Inhibition of hepatitis c virus replication by peroxidation of arachidonate and restoration by vitamin e. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18666–18670. [Google Scholar] [CrossRef]
- Trujillo–Murillo, K.; Alvarez–Martinez, O.; Garza–Rodriguez, L.; Martinez–Rodriguez, H.; Bosques–Padilla, F.; Ramos–Jimenez, J.; Barrera–Saldana, H.; Rincon–Sanchez, A.R.; Rivas–Estilla, A.M. Additive effect of ethanol and hcv subgenomic replicon expression on cox–2 protein levels and activity. J. Viral Hepat. 2007, 14, 608–617. [Google Scholar] [CrossRef]
- McCartney, E.M.; Semendric, L.; Helbig, K.J.; Hinze, S.; Jones, B.; Weinman, S.A.; Beard, M.R. Alcohol metabolism increases the replication of hepatitis c virus and attenuates the antiviral action of interferon. J. Infect. Dis. 2008, 198, 1766–1775. [Google Scholar] [CrossRef]
- von Herbay, A.; Stahl, W.; Niederau, C.; Sies, H. Vitamin e improves the aminotransferase status of patients suffering from viral hepatitis c: A randomized, double–blind, placebo–controlled study. Free Radical Res. 1997, 27, 599–605. [Google Scholar] [CrossRef]
- Par, A.; Roth, E.; Miseta, A.; Hegedus, G.; Par, G.; Hunyady, B.; Vincze, A. Effects of supplementation with the antioxidant flavonoid, silymarin, in chronic hepatitis c patients treated with peg–interferon + ribavirin. A placebo–controlled double blind study. Orv Hetil 2009, 150, 73–79. [Google Scholar] [CrossRef]
- Farias, M.S.; Budni, P.; Ribeiro, C.M.; Parisotto, E.B.; Santos, C.E.; Dias, J.F.; Dalmarco, E.M.; Frode, T.S.; Pedrosa, R.C.; Wilhelm Filho, D. Antioxidant supplementation attenuates oxidative stress in chronic hepatitis c patients. Gastroenterol. Hepatol. 2012, 35, 386–394. [Google Scholar] [CrossRef]
- Melhem, A.; Stern, M.; Shibolet, O.; Israeli, E.; Ackerman, Z.; Pappo, O.; Hemed, N.; Rowe, M.; Ohana, H.; Zabrecky, G.; et al. Treatment of chronic hepatitis c virus infection via antioxidants: Results of a phase i clinical trial. J. Clin. Gastroenterol. 2005, 39, 737–742. [Google Scholar] [CrossRef]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.; Murphy, M.P. The mitochondria–targeted anti–oxidant mitoquinone decreases liver damage in a phase ii study of hepatitis c patients. Liver Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef]
- Choi, J.; Lee, K.J.; Zheng, Y.; Yamaga, A.K.; Lai, M.M.; Ou, J.H. Reactive oxygen species suppress hepatitis c virus rna replication in human hepatoma cells. Hepatology 2004, 39, 81–89. [Google Scholar] [CrossRef]
- Rieusset, J.; Fauconnier, J.; Paillard, M.; Belaidi, E.; Tubbs, E.; Chauvin, M.A.; Durand, A.; Bravard, A.; Teixeira, G.; Bartosch, B.; et al. Disruption of cyclophilin d–mediated calcium transfer from the er to mitochondria contributes to hepatic er stress and insulin resistance. Hepatology 2012. [Google Scholar]
- Distelhorst, C.W.; Bootman, M.D. Bcl–2 interaction with the inositol 1,4,5–trisphosphate receptor: Role in ca(2+) signaling and disease. Cell Calcium 2011, 50, 234–241. [Google Scholar] [CrossRef]
- Benali–Furet, N.L.; Chami, M.; Houel, L.; De Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; et al. Hepatitis c virus core triggers apoptosis in liver cells by inducing er stress and er calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar]
- Calabrese, F.; Pontisso, P.; Pettenazzo, E.; Benvegnu, L.; Vario, A.; Chemello, L.; Alberti, A.; Valente, M. Liver cell apoptosis in chronic hepatitis c correlates with histological but not biochemical activity or serum hcv–rna levels. Hepatology 2000, 31, 1153–1159. [Google Scholar] [CrossRef]
- Bantel, H.; Lugering, A.; Heidemann, J.; Volkmann, X.; Poremba, C.; Strassburg, C.P.; Manns, M.P.; Schulze–Osthoff, K. Detection of apoptotic caspase activation in sera from patients with chronic hcv infection is associated with fibrotic liver injury. Hepatology 2004, 40, 1078–1087. [Google Scholar] [CrossRef]
- Kronenberger, B.; Ruster, B.; Lee, J.H.; Sarrazin, C.; Roth, W.K.; Herrmann, G.; Zeuzem, S. Hepatocellular proliferation in patients with chronic hepatitis c and persistently normal or abnormal aminotransferase levels. J. Hepatol. 2000, 33, 640–647. [Google Scholar] [CrossRef]
- Lau, J.Y.; Xie, X.; Lai, M.M.; Wu, P.C. Apoptosis and viral hepatitis. Semin. Liver Dis. 1998, 18, 169–176. [Google Scholar] [CrossRef]
- Canbay, A.; Friedman, S.; Gores, G.J. Apoptosis: The nexus of liver injury and fibrosis. Hepatology 2004, 39, 273–278. [Google Scholar] [CrossRef]
- Hiramatsu, N.; Hayashi, N.; Haruna, Y.; Kasahara, A.; Fusamoto, H.; Mori, C.; Fuke, I.; Okayama, H.; Kamada, T. Immunohistochemical detection of hepatitis c virus–infected hepatocytes in chronic liver disease with monoclonal antibodies to core, envelope and ns3 regions of the hepatitis c virus genome. Hepatology 1992, 16, 306–311. [Google Scholar] [CrossRef]
- Liang, Y.; Shilagard, T.; Xiao, S.Y.; Snyder, N.; Lau, D.; Cicalese, L.; Weiss, H.; Vargas, G.; Lemon, S.M. Visualizing hepatitis c virus infections in human liver by two–photon microscopy. Gastroenterology 2009, 137, 1448–1458. [Google Scholar] [CrossRef]
- Moorman, J.P.; Prayther, D.; McVay, D.; Hahn, Y.S.; Hahn, C.S. The c–terminal region of hepatitis c core protein is required for fas–ligand independent apoptosis in jurkat cells by facilitating fas oligomerization. Virology 2003, 312, 320–329. [Google Scholar] [CrossRef]
- Lee, S.K.; Park, S.O.; Joe, C.O.; Kim, Y.S. Interaction of hcv core protein with 14–3–3epsilon protein releases bax to activate apoptosis. Biochem. Biophys. Res. Commun. 2007, 352, 756–762. [Google Scholar] [CrossRef]
- Mohd–Ismail, N.K.; Deng, L.; Sukumaran, S.K.; Yu, V.C.; Hotta, H.; Tan, Y.J. The hepatitis c virus core protein contains a bh3 domain that regulates apoptosis through specific interaction with human mcl–1. J. Virol. 2009, 83, 9993–10006. [Google Scholar] [CrossRef]
- Goh, P.Y.; Tan, Y.J.; Lim, S.P.; Lim, S.G.; Tan, Y.H.; Hong, W.J. The hepatitis c virus core protein interacts with ns5a and activates its caspase–mediated proteolytic cleavage. Virology 2001, 290, 224–236. [Google Scholar] [CrossRef]
- Sacco, R.; Tsutsumi, T.; Suzuki, R.; Otsuka, M.; Aizaki, H.; Sakamoto, S.; Matsuda, M.; Seki, N.; Matsuura, Y.; Miyamura, T.; et al. Antiapoptotic regulation by hepatitis c virus core protein through up–regulation of inhibitor of caspase–activated dnase. Virology 2003, 317, 24–35. [Google Scholar] [CrossRef]
- Chou, A.H.; Tsai, H.F.; Wu, Y.Y.; Hu, C.Y.; Hwang, L.H.; Hsu, P.I.; Hsu, P.N. Hepatitis c virus core protein modulates trail–mediated apoptosis by enhancing bid cleavage and activation of mitochondria apoptosis signaling pathway. J. Immunol. 2005, 174, 2160–2166. [Google Scholar]
- Berg, C.P.; Schlosser, S.F.; Neukirchen, D.K.; Papadakis, C.; Gregor, M.; Wesselborg, S.; Stein, G.M. Hepatitis c virus core protein induces apoptosis–like caspase independent cell death. Virol. J. 2009, 6, 213. [Google Scholar] [CrossRef]
- Ciccaglione, A.R.; Marcantonio, C.; Tritarelli, E.; Equestre, M.; Magurano, F.; Costantino, A.; Nicoletti, L.; Rapicetta, M. The transmembrane domain of hepatitis c virus e1 glycoprotein induces cell death. Virus Res. 2004, 104, 1–9. [Google Scholar] [CrossRef]
- Ciccaglione, A.R.; Marcantonio, C.; Costantino, A.; Equestre, M.; Rapicetta, M. Expression of hcv e1 protein in baculovirus–infected cells: Effects on cell viability and apoptosis induction. Intervirology 2003, 46, 121–126. [Google Scholar] [CrossRef]
- Chiou, H.L.; Hsieh, Y.S.; Hsieh, M.R.; Chen, T.Y. Hcv e2 may induce apoptosis of huh–7 cells via a mitochondrial–related caspase pathway. Biochem. Biophys. Res. Commun. 2006, 345, 453–458. [Google Scholar] [CrossRef]
- Erdtmann, L.; Franck, N.; Lerat, H.; Le Seyec, J.; Gilot, D.; Cannie, I.; Gripon, P.; Hibner, U.; Guguen–Guillouzo, C. The hepatitis c virus ns2 protein is an inhibitor of cide–b–induced apoptosis. J. Biol. Chem. 2003, 278, 18256–18264. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the rig–i antiviral pathway and is targeted by hepatitis c virus. Nature 2005, 437, 1167–1172. [Google Scholar]
- Prikhod'ko, E.A.; Prikhod'ko, G.G.; Siegel, R.M.; Thompson, P.; Major, M.E.; Cohen, J.I. The ns3 protein of hepatitis c virus induces caspase–8–mediated apoptosis independent of its protease or helicase activities. Virology 2004, 329, 53–67. [Google Scholar] [CrossRef]
- Zhao, P.; Han, T.; Guo, J.J.; Zhu, S.L.; Wang, J.; Ao, F.; Jing, M.Z.; She, Y.L.; Wu, Z.H.; Ye, L.B. Hcv ns4b induces apoptosis through the mitochondrial death pathway. Virus Res. 2012, 169, 1–7. [Google Scholar] [CrossRef]
- Zhou, Y.; Gong, G.Z. Hepatitis c virus ns5a protein upregulates survivin gene expression. Zhonghua Gan Zang Bing Za Zhi 2006, 14, 414–417. [Google Scholar]
- Lin, L.Y.; Li, S.C.; Lu, S.L. Hepatitis c virus nonstructural 5a protein inhibits tumor necrosis factor alpha mediated apoptosis of hepg2 cells. Zhonghua Nei Ke Za Zhi 2003, 42, 392–395. [Google Scholar]
- Lan, K.H.; Sheu, M.L.; Hwang, S.J.; Yen, S.H.; Chen, S.Y.; Wu, J.C.; Wang, Y.J.; Kato, N.; Omata, M.; Chang, F.Y.; et al. Hcv ns5a interacts with p53 and inhibits p53–mediated apoptosis. Oncogene 2002, 21, 4801–4811. [Google Scholar] [CrossRef]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The hepatitis c virus ns5a protein activates a phosphoinositide 3–kinase–dependent survival signaling cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar]
- Chung, Y.L.; Sheu, M.L.; Yen, S.H. Hepatitis c virus ns5a as a potential viral bcl–2 homologue interacts with bax and inhibits apoptosis in hepatocellular carcinoma. Int. J. Cancer 2003, 107, 65–73. [Google Scholar] [CrossRef]
- Quarato, G.; D'Aprile, A.; Gavillet, B.; Vuagniaux, G.; Moradpour, D.; Capitanio, N.; Piccoli, C. The cyclophilin inhibitor alisporivir prevents hepatitis c virus– mediated mitochondrial dysfunction. Hepatology 2012, 55, 1333–1343. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brault, C.; Levy, P.L.; Bartosch, B. Hepatitis C Virus-Induced Mitochondrial Dysfunctions. Viruses 2013, 5, 954-980. https://doi.org/10.3390/v5030954
Brault C, Levy PL, Bartosch B. Hepatitis C Virus-Induced Mitochondrial Dysfunctions. Viruses. 2013; 5(3):954-980. https://doi.org/10.3390/v5030954
Chicago/Turabian StyleBrault, Charlène, Pierre L. Levy, and Birke Bartosch. 2013. "Hepatitis C Virus-Induced Mitochondrial Dysfunctions" Viruses 5, no. 3: 954-980. https://doi.org/10.3390/v5030954
APA StyleBrault, C., Levy, P. L., & Bartosch, B. (2013). Hepatitis C Virus-Induced Mitochondrial Dysfunctions. Viruses, 5(3), 954-980. https://doi.org/10.3390/v5030954