
Mutagenic Effects of Ribavirin on Hepatitis E Virus—Viral Extinction versus Selection of Fitness-Enhancing Mutations




{kind=link}
{kind=link}
Abstract
:1. Introduction
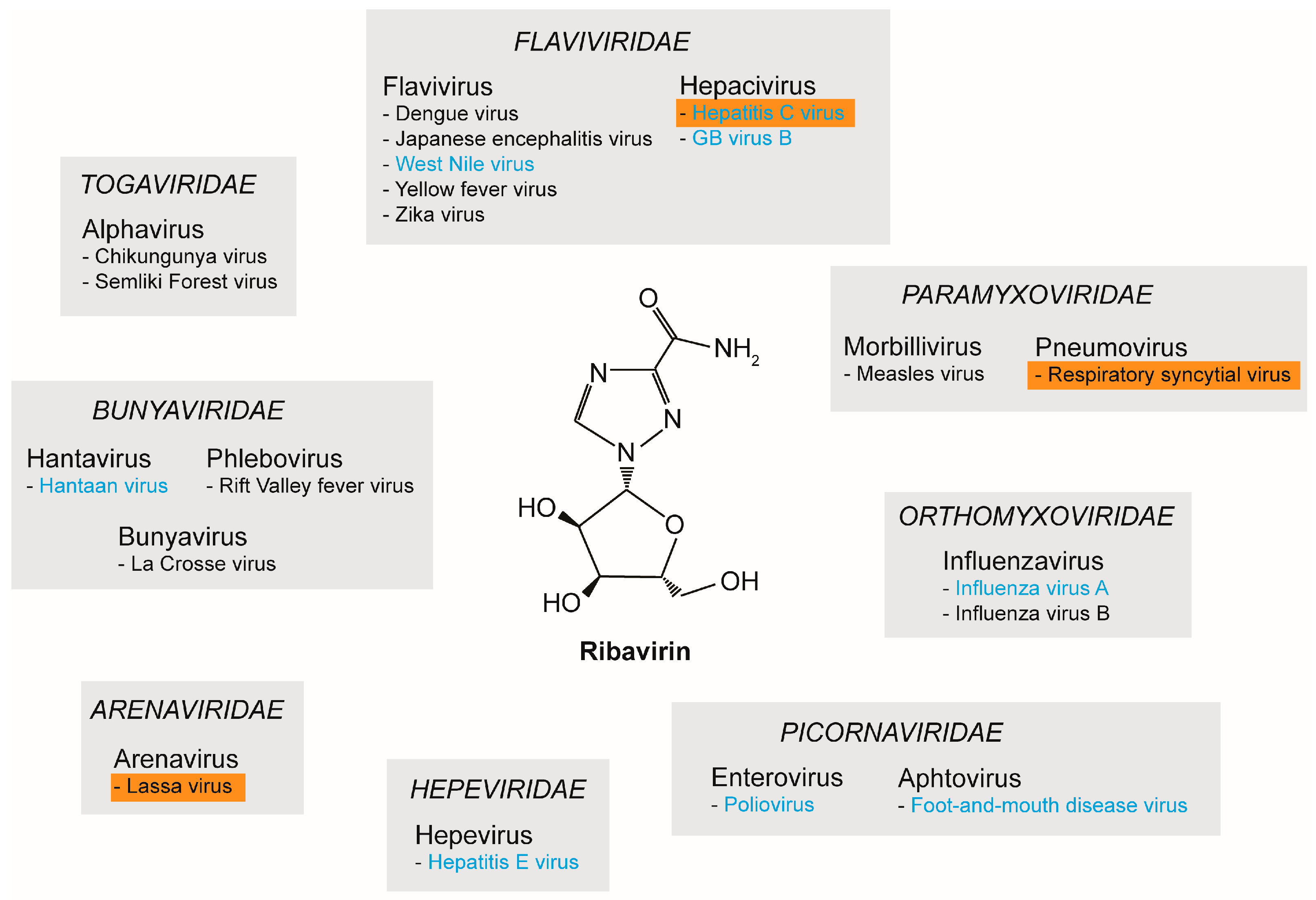
2. RNA Viruses and Ribavirin
3. Multiple Modes of Action for Ribavirin
4. Hepatitis E Virus as Intra-Host Viral Populations
5. Hepatitis E Virus and Mechanisms of Ribavirin Action
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Reyes, G.R.; Yarbough, P.O.; Tam, A.W.; Purdy, M.A.; Huang, C.C.; Kim, J.S.P.; Bradley, D.W.; Fry, K.E. Hepatitis-E Virus (HEV)—The Novel Agent Responsible for Enterically Transmitted Non-A, Non-B Hepatitis. Gastroenterol. Jpn. 1991, 26, 142–147. [Google Scholar] [PubMed]
- Viswanathan, R. Infectious hepatitis in Delhi (1955–1956): A critical study-epidemiology. 1957. Natl. Med. J. India 2013, 26, 362–377. [Google Scholar] [PubMed]
- Debing, Y.; Moradpour, D.; Neyts, J.; Gouttenoire, J. Update on hepatitis E virology: Implications for clinical practice. J. Hepatol. 2016, 65, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Simmonds, P.; International Committee on Taxonomy of Viruses Hepeviridae Study Group; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; van der Poel, W.H.; Purdy, M.A. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95 Pt 10, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Blum, H.E. History and Global Burden of Viral Hepatitis. Dig. Dis. 2016, 34, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H.; Pischke, S.; Manns, M.P. Pathogenesis and treatment of hepatitis E virus infection. Gastroenterology 2012, 142, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Sahai, S.; Mishra, V.; Ganga, D.; Jatav, O.P. Viral Hepatitis in Pregnancy—A study of its Effect on Maternal and Foetal Outcome. J. Assoc. Physicians India 2015, 63, 28–33. [Google Scholar] [PubMed]
- Debes, J.D.; Pisano, M.B.; Lotto, M.; Re, V. Corrigendum to “Hepatitis E virus infection in the HIV-positive patient”. J. Clin. Virol. 2016, 82, 181–182. [Google Scholar] [CrossRef] [PubMed]
- Debes, J.D.; Pisano, M.B.; Lotto, M.; Re, V. Hepatitis E virus infection in the HIV-positive patient. J. Clin. Virol. 2016, 80, 102–106. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Hepatitis E (Fact Sheet). Available online: http://www.who.int/mediacentre/factsheets/fs280/en/ (accessed on 25 July 2016).
- Marano, G.; Vaglio, S.; Pupella, S.; Facco, G.; Bianchi, M.; Calizzani, G.; Candura, F.; Catalano, L.; Farina, B.; Lanzoni, M.; et al. Hepatitis E: An old infection with new implications. Blood Transfus. 2015, 13, 6–17. [Google Scholar] [PubMed]
- Pischke, S.; Behrendt, P.; Bock, C.T.; Jilg, W.; Manns, M.P.; Wedemeyer, H. Hepatitis E in Germany—An under-reported infectious disease. Dtsch. Arzteblatt Int. 2014, 111, 577–583. [Google Scholar]
- Perrin, H.B.; Cintas, P.; Abravanel, F.; Gerolami, R.; d’Alteroche, L.; Raynal, J.N.; Alric, L.; Dupuis, E.; Prudhomme, L.; Vaucher, E.; et al. Neurologic Disorders in Immunocompetent Patients with Autochthonous Acute Hepatitis E. Emerg. Infect. Dis. 2015, 21, 1928–1934. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Kamar, N.; van Eijk, J.J.J.; Mclean, B.N.; Cintas, P.; Bendall, R.P.; Jacobs, B.C. Hepatitis E virus and neurological injury. Nat. Rev. Neurol. 2016, 12, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, B.; van der Eijk, A.A.; Pas, S.D.; Hunter, J.G.; Madden, R.G.; Tio-Gillen, A.P.; Dalton, H.R.; Jacobs, B.C. Guillain-Barre syndrome associated with preceding hepatitis E virus infection. Neurology 2014, 82, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Soomro, M.H.; She, R.; Yang, Y.; Wang, T.; Wu, Q.; Li, H.; Hao, W. Evidence of Hepatitis E virus breaking through the blood-brain barrier and replicating in the central nervous system. J. Viral. Hepat. 2016. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Wicki, B.; Tsouni, P.; Cunningham, S.; Doerig, C.; Zanetti, G.; Aubert, V.; Sahli, R.; Moradpour, D.; Kuntzer, T. Hepatitis E virus infection as a direct cause of neuralgic amyotrophy. Muscle Nerve 2016, 54, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Loly, J.P.; Rikir, E.; Seivert, M.; Legros, E.; Defrance, P.; Belaiche, J.; Moonen, G.; Delwaide, J. Guillain-Barre syndrome following hepatitis E. World J. Gastroenterol. 2009, 15, 1645–1647. [Google Scholar] [CrossRef] [PubMed]
- Drave, S.A.; Debing, Y.; Walter, S.; Todt, D.; Engelmann, M.; Friesland, M.; Wedemeyer, H.; Neyts, J.; Behrendt, P.; Steinmann, E. Extra-hepatic replication and infection of hepatitis E virus in neuronal-derived cells. J. Viral. Hepat. 2016, 23, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Feigelstock, D.A.; Mihalik, K.B.; Feinstone, S.M. Selection of hepatitis C virus resistant to ribavirin. Virol. J. 2011, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Sierra, M.; Airaksinen, A.; Gonzalez-Lopez, C.; Agudo, R.; Arias, A.; Domingo, E. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: Implications for error catastrophe. J. Virol. 2007, 81, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef] [PubMed]
- Scheidel, L.M.; Durbin, R.K.; Stollar, V. Sindbis virus mutants resistant to mycophenolic acid and ribavirin. Virology 1987, 158, 1–7. [Google Scholar] [CrossRef]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A Mutation in the hepatitis E virus RNA polymerase promotes its replication and associates with ribavirin treatment failure in organ transplant recipients. Gastroenterology 2014, 147, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Ramiere, C.; Dallmeier, K.; Piorkowski, G.; Trabaud, M.A.; Lebosse, F.; Scholtes, C.; Roche, M.; Legras-Lachuer, C.; de Lamballerie, X.; et al. Hepatitis E virus mutations associated with ribavirin treatment failure result in altered viral fitness and ribavirin sensitivity. J. Hepatol. 2016, 65, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; Gisa, A.; Radonic, A.; Nitsche, A.; Behrendt, P.; Suneetha, P.V.; Pischke, S.; Bremer, B.; Brown, R.J.; Manns, M.P.; et al. In vivo evidence for ribavirin-induced mutagenesis of the hepatitis E virus genome. Gut 2016, 65, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Sidwell, R.W.; Huffman, J.H.; Khare, G.P.; Allen, L.B.; Witkowski, J.T.; Robins, R.K. Broad-spectrum antiviral activity of Virazole: 1-Beta-d-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 1972, 177, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Andino, R. Ribavirin and lethal mutagenesis of poliovirus: Molecular mechanisms, resistance and biological implications. Virus Res. 2005, 107, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Graci, J.D.; Cameron, C.E. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 2006, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Snell, N.J. Ribavirin—Current status of a broad spectrum antiviral agent. Expert Opin. Pharmacother. 2001, 2, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Crance, J.M.; Scaramozzino, N.; Jouan, A.; Garin, D. Interferon, ribavirin, 6-azauridine and glycyrrhizin: Antiviral compounds active against pathogenic flaviviruses. Antivir. Res. 2003, 58, 73–79. [Google Scholar] [CrossRef]
- Monath, T.P. Treatment of yellow fever. Antivir. Res. 2008, 78, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Julander, J.; Shafer, K.; Morrey, J.; Blatt, L.; Sidwell, R. Comparison of the inhibitory effects of ribavirin and interferon alfacon 1 on a yellow fever virus infection in Syrian golden hamsters. Antivir. Res. 2006, 70, A82–A83. [Google Scholar]
- Huggins, J.W. Prospects for treatment of viral hemorrhagic fevers with ribavirin, a broad-spectrum antiviral drug. Rev. Infect. Dis. 1989, 11 (Suppl. 4), S750–S761. [Google Scholar] [CrossRef] [PubMed]
- Andrei, G.; de Clercq, E. Molecular approaches for the treatment of hemorrhagic fever virus infections. Antivir. Res. 1993, 22, 45–75. [Google Scholar] [CrossRef]
- Burt, F.J.; Rolph, M.S.; Rulli, N.E.; Mahalingam, S.; Heise, M.T. Chikungunya: A re-emerging virus. Lancet 2012, 379, 662–671. [Google Scholar] [CrossRef]
- Briolant, S.; Garin, D.; Scaramozzino, N.; Jouan, A.; Crance, J.M. In vitro inhibition of Chikungunya and Semliki Forest viruses replication by antiviral compounds: Synergistic effect of interferon-alpha and ribavirin combination. Antivir. Res. 2004, 61, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, A.; Pariente, N.; Menendez-Arias, L.; Domingo, E. Curing of foot-and-mouth disease virus from persistently infected cells by ribavirin involves enhanced mutagenesis. Virology 2003, 311, 339–349. [Google Scholar] [CrossRef]
- Pariente, N.; Sierra, S.; Airaksinen, A. Action of mutagenic agents and antiviral inhibitors on foot-and-mouth disease virus. Virus Res. 2005, 107, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [PubMed]
- Stein, D.S.; Creticos, C.M.; Jackson, G.G.; Bernstein, J.M.; Hayden, F.G.; Schiff, G.M.; Bernstein, D.I. Oral ribavirin treatment of influenza A and B. Antimicrob. Agents Chemother. 1987, 31, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Knight, V.; McClung, H.W.; Wilson, S.Z.; Waters, B.K.; Quarles, J.M.; Cameron, R.W.; Greggs, S.E.; Zerwas, J.M.; Couch, R.B. Ribavirin small-particle aerosol treatment of influenza. Lancet 1981, 2, 945–949. [Google Scholar] [CrossRef]
- Debing, Y.; Jochmans, D.; Neyts, J. Intervention strategies for emerging viruses: Use of antivirals. Curr. Opin. Virol. 2013, 3, 217–224. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Scribner, C.L.; Craven, R.B.; Johnson, K.M.; Elliott, L.H.; Belmont-Williams, R. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986, 314, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H.; Goba, A.; Chu, M.; Roth, C.; Healing, T.; Marx, A.; Fair, J.; Guttieri, M.C.; Ferro, P.; Imes, T.; et al. New opportunities for field research on the pathogenesis and treatment of Lassa fever. Antivir. Res. 2008, 78, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Picardi, A.; Gentilucci, U.V.; Zardi, E.M.; D’Avola, D.; Amoroso, A.; Afeltra, A. The role of ribavirin in the combination therapy of hepatitis C virus infection. Curr. Pharm. Des. 2004, 10, 2081–2092. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. Mechanisms of antiviral treatment efficacy and failure in chronic hepatitis C. Antivir. Res. 2003, 59, 1–11. [Google Scholar] [CrossRef]
- Cummings, K.J.; Lee, S.M.; West, E.S.; Cid-Ruzafa, J.; Fein, S.G.; Aoki, Y.; Sulkowski, M.S.; Goodman, S.N. Interferon and ribavirin vs. interferon alone in the re-treatment of chronic hepatitis C previously nonresponsive to interferon: A meta-analysis of randomized trials. JAMA 2001, 285, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Mejias, A.; Ramilo, O. New options in the treatment of respiratory syncytial virus disease. J. Infect. 2015, 71 (Suppl. 1), S80–S87. [Google Scholar] [CrossRef] [PubMed]
- Ventre, K.; Randolph, A.G. Ribavirin for respiratory syncytial virus infection of the lower respiratory tract in infants and young children. Cochrane Database Syst. Rev. 2007. [Google Scholar] [CrossRef]
- Paeshuyse, J.; Dallmeier, K.; Neyts, J. Ribavirin for the treatment of chronic hepatitis C virus infection: A review of the proposed mechanisms of action. Curr. Opin. Virol. 2011, 1, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Hultgren, C.; Milich, D.R.; Weiland, O.; Sallberg, M. The antiviral compound ribavirin modulates the T helper (Th) 1/Th2 subset balance in hepatitis B and C virus-specific immune responses. J. Gen. Virol. 1998, 79 Pt 10, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Sad, S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 1996, 17, 138–146. [Google Scholar] [CrossRef]
- Edell, D.; Bruce, E.; Hale, K.; Edell, D.; Khoshoo, V. Reduced long-term respiratory morbidity after treatment of respiratory syncytial virus bronchiolitis with ribavirin in previously healthy infants: A preliminary report. Pediatr. Pulmonol. 1998, 25, 154–158. [Google Scholar] [CrossRef]
- Streeter, D.G.; Witkowski, J.T.; Khare, G.P.; Sidwell, R.W.; Bauer, R.J.; Robins, R.K.; Simon, L.N. Mechanism of action of 1-β-d-ribofuranosyl-1,2,4-triazole-3-carboxamide (Virazole), a new broad-spectrum antiviral agent. Proc. Natl. Acad. Sci. USA 1973, 70, 1174–1178. [Google Scholar] [CrossRef] [PubMed]
- Leyssen, P.; Balzarini, J.; de Clercq, E.; Neyts, J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J. Virol. 2005, 79, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Chavez, D.; Guerra, B.; Lau, J.Y.; Hong, Z.; Brasky, K.M.; Beames, B. Ribavirin induces error-prone replication of GB virus B in primary tamarin hepatocytes. J. Virol. 2001, 75, 8074–8081. [Google Scholar] [CrossRef] [PubMed]
- Olschlager, S.; Neyts, J.; Gunther, S. Depletion of GTP pool is not the predominant mechanism by which ribavirin exerts its antiviral effect on Lassa virus. Antivir. Res. 2011, 91, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chung, D.H.; Chu, Y.K.; Jonsson, C.B.; Parker, W.B. Activity of ribavirin against Hantaan virus correlates with production of ribavirin-5′-triphosphate, not with inhibition of IMP dehydrogenase. Antimicrob. Agents Chemother. 2007, 51, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Wray, S.K.; Gilbert, B.E.; Noall, M.W.; Knight, V. Mode of action of ribavirin: Effect of nucleotide pool alterations on influenza virus ribonucleoprotein synthesis. Antivir. Res. 1985, 5, 29–37. [Google Scholar] [CrossRef]
- Knight, V.; Wilson, S.Z.; Alling, D.W.; Moore, R.V.; Longoria, R.M. Lack of interference of guanosine with ribavirin aerosol treatment of influenza A infection in mice. Antimicrob. Agents Chemother. 1981, 20, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Rotman, Y.; Noureddin, M.; Feld, J.J.; Guedj, J.; Witthaus, M.; Han, H.; Park, Y.J.; Park, S.H.; Heller, T.; Ghany, M.G.; et al. Effect of ribavirin on viral kinetics and liver gene expression in chronic hepatitis C. Gut 2014, 63, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Tokumoto, Y.; Hiasa, Y.; Uesugi, K.; Watanabe, T.; Mashiba, T.; Abe, M.; Kumagi, T.; Ikeda, Y.; Matsuura, B.; Onji, M. Ribavirin regulates hepatitis C virus replication through enhancing interferon-stimulated genes and interleukin 8. J. Infect. Dis. 2012, 205, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Lutchman, G.A.; Heller, T.; Hara, K.; Pfeiffer, J.K.; Leff, R.D.; Meek, C.; Rivera, M.; Ko, M.; Koh, C.; et al. Ribavirin improves early responses to peginterferon through improved interferon signaling. Gastroenterology 2010, 139, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Nanda, S.; Huang, Y.; Chen, W.; Cam, M.; Pusek, S.N.; Schweigler, L.M.; Theodore, D.; Zacks, S.L.; Liang, T.J.; et al. Hepatic gene expression during treatment with peginterferon and ribavirin: Identifying molecular pathways for treatment response. Hepatology 2007, 46, 1548–1563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jamaluddin, M.; Wang, S.; Tian, B.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Ribavirin treatment up-regulates antiviral gene expression via the interferon-stimulated response element in respiratory syncytial virus-infected epithelial cells. J. Virol. 2003, 77, 5933–5947. [Google Scholar] [CrossRef] [PubMed]
- Su, W.C.; Liu, W.L.; Cheng, C.W.; Chou, Y.B.; Hung, K.H.; Huang, W.H.; Wu, C.L.; Li, Y.T.; Shiau, A.L.; Lai, M.Y. Ribavirin enhances interferon signaling via stimulation of mTOR and p53 activities. FEBS Lett. 2009, 583, 2793–2798. [Google Scholar] [CrossRef] [PubMed]
- Testoni, B.; Durantel, D.; Lebosse, F.; Fresquet, J.; Helle, F.; Negro, F.; Donato, M.F.; Levrero, M.; Zoulim, F. Ribavirin restores IFNalpha responsiveness in HCV-infected livers by epigenetic remodelling at interferon stimulated genes. Gut 2016, 65, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Bisaillon, M.; Lemay, G. Viral and cellular enzymes involved in synthesis of mRNA cap structure. Virology 1997, 236, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, D.; Egloff, M.P.; Mulard, L.; Guerreiro, C.; Romette, J.L.; Canard, B. A structural basis for the inhibition of the NS5 dengue virus mRNA 2′-O-methyltransferase domain by ribavirin 5′-triphosphate. J. Biol. Chem. 2004, 279, 35638–35643. [Google Scholar] [CrossRef] [PubMed]
- Bougie, I.; Bisaillon, M. The broad spectrum antiviral nucleoside ribavirin as a substrate for a viral RNA capping enzyme. J. Biol. Chem. 2004, 279, 22124–22130. [Google Scholar] [CrossRef] [PubMed]
- Goswami, B.B.; Borek, E.; Sharma, O.K.; Fujitaki, J.; Smith, R.A. The broad spectrum antiviral agent ribavirin inhibits capping of mRNA. Biochem. Biophys. Res. Commun. 1979, 89, 830–836. [Google Scholar] [CrossRef]
- Yan, Y.; Svitkin, Y.; Lee, J.M.; Bisaillon, M.; Pelletier, J. Ribavirin is not a functional mimic of the 7-methyl guanosine mRNA cap. RNA 2005, 11, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Westman, B.; Beeren, L.; Grudzien, E.; Stepinski, J.; Worch, R.; Zuberek, J.; Jemielity, J.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E.; et al. The antiviral drug ribavirin does not mimic the 7-methylguanosine moiety of the mRNA cap structure in vitro. RNA 2005, 11, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Ghany, M.G.; Liang, T.J. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir. Chem. Chemother. 2013, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, B.; Helgstrand, E.; Johansson, N.G.; Larsson, A.; Misiorny, A.; Noren, J.O.; Philipson, L.; Stenberg, K.; Stening, G.; Stridh, S.; et al. Inhibition of influenza virus ribonucleic acid polymerase by ribavirin triphosphate. Antimicrob. Agents Chemother. 1977, 11, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Toltzis, P.; O’Connell, K.; Patterson, J.L. Effect of phosphorylated ribavirin on vesicular stomatitis virus transcription. Antimicrob. Agents Chemother. 1988, 32, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Rankin, J.T., Jr.; Eppes, S.B.; Antczak, J.B.; Joklik, W.K. Studies on the mechanism of the antiviral activity of ribavirin against reovirus. Virology 1989, 168, 147–158. [Google Scholar] [CrossRef]
- Lau, J.Y.; Tam, R.C.; Liang, T.J.; Hong, Z. Mechanism of action of ribavirin in the combination treatment of chronic HCV infection. Hepatology 2002, 35, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.T.; Gale, M., Jr.; Polyak, S.J.; Lemon, S.M.; Liang, T.J.; Hoofnagle, J.H. Mechanisms of action of interferon and ribavirin in chronic hepatitis C: Summary of a workshop. Hepatology 2008, 47, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M.; Dahari, H.; Neumann, A.U.; Hezode, C.; Germanidis, G.; Lonjon, I.; Castera, L.; Dhumeaux, D. Antiviral action of ribavirin in chronic hepatitis C. Gastroenterology 2004, 126, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Schuster, P. What Is a Quasispecies? Historical Origins and Current Scope. Curr. Top. Microbiol. Immunol. 2016, 392, 1–22. [Google Scholar] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C. The RNA virus quasispecies: Fact or fiction? J. Mol. Biol. 2010, 400, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.M.; Hiasa, Y.; He, W.; Terella, A.; Schmidt, E.V.; Chung, R.T. Viral RNA mutations are region specific and increased by ribavirin in a full-length hepatitis C virus replication system. J. Virol. 2002, 76, 8505–8517. [Google Scholar] [CrossRef] [PubMed]
- Severson, W.E.; Schmaljohn, C.S.; Javadian, A.; Jonsson, C.B. Ribavirin causes error catastrophe during Hantaan virus replication. J. Virol. 2003, 77, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Lhomme, S.; Abravanel, F.; Marion, O.; Peron, J.M.; Alric, L.; Izopet, J. Treatment of HEV Infection in Patients with a Solid-Organ Transplant and Chronic Hepatitis. Viruses 2016, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Kamar, N. Treatment of hepatitis E virus. Curr. Opin. Infect. Dis. 2016, 29. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M.; Schuster, P. The hypercycle. A principle of natural self-organization. Part A: Emergence of the hypercycle. Naturwissenschaften 1977, 64, 541–565. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Davila, M.; Ortin, J. Nucleotide sequence heterogeneity of the RNA from a natural population of foot-and-mouth-disease virus. Gene 1980, 11, 333–346. [Google Scholar] [CrossRef]
- Sobrino, F.; Davila, M.; Ortin, J.; Domingo, E. Multiple genetic variants arise in the course of replication of foot-and-mouth disease virus in cell culture. Virology 1983, 128, 310–318. [Google Scholar] [CrossRef]
- Spindler, K.R.; Horodyski, F.M.; Holland, J.J. High multiplicities of infection favor rapid and random evolution of vesicular stomatitis virus. Virology 1982, 119, 96–108. [Google Scholar] [CrossRef]
- Holland, J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; VandePol, S. Rapid evolution of RNA genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef]
- Domingo, E.; Wain-Hobson, S. The 30th anniversary of quasispecies. Meeting on ‘Quasispecies: Past, present and future’. EMBO Rep. 2009, 10, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.U.; Lam, N.P.; Dahari, H.; Gretch, D.R.; Wiley, T.E.; Layden, T.J.; Perelson, A.S. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 1998, 282, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Bonhoeffer, S.; Hill, A.M.; Boehme, R.; Thomas, H.C.; McDade, H. Viral dynamics in hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 1996, 93, 4398–4402. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.D.; Carpenter, C.D.; Simon, A.E. A novel 3′-end repair mechanism in an RNA virus. Proc. Natl. Acad. Sci. USA 1997, 94, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3′→5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.; Denison, M.R. Implications of altered replication fidelity on the evolution and pathogenesis of coronaviruses. Curr. Opin. Virol. 2012, 2, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Leveque, V.; Ma, H.; Johnson, K.A.; Klumpp, K. NTP-mediated nucleotide excision activity of hepatitis C virus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2013, 110, E348–E357. [Google Scholar] [CrossRef] [PubMed]
- Deval, J.; Powdrill, M.H.; D’Abramo, C.M.; Cellai, L.; Gotte, M. Pyrophosphorolytic excision of nonobligate chain terminators by hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 2007, 51, 2920–2928. [Google Scholar] [CrossRef] [PubMed]
- Grandadam, M.; Tebbal, S.; Caron, M.; Siriwardana, M.; Larouze, B.; Koeck, J.L.; Buisson, Y.; Enouf, V.; Nicand, E. Evidence for hepatitis E virus quasispecies. J. Gen. Virol. 2004, 85 Pt 11, 3189–3194. [Google Scholar] [CrossRef] [PubMed]
- Cerni, S.; Prpic, J.; Jemersic, L.; Skoric, D. The application of single strand conformation polymorphism (SSCP) analysis in determining Hepatitis E virus intra-host diversity. J. Virol. Methods 2015, 221, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Rostaing, L.; Kamar, N.; Izopet, J. Hepatitis E virus quasispecies and the outcome of acute hepatitis E in solid-organ transplant patients. J. Virol. 2012, 86, 10006–10014. [Google Scholar] [CrossRef] [PubMed]
- Tejero, H.; Montero, F.; Nuno, J.C. Theories of Lethal Mutagenesis: From Error Catastrophe to Lethal Defection. Curr. Top. Microbiol. Immunol. 2016, 392, 161–179. [Google Scholar] [PubMed]
- Perales, C.; Agudo, R.; Domingo, E. Counteracting quasispecies adaptability: Extinction of a ribavirin-resistant virus mutant by an alternative mutagenic treatment. PLoS ONE 2009, 4, e5554. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Agudo, R.; Tejero, H.; Manrubia, S.C.; Domingo, E. Potential benefits of sequential inhibitor-mutagen treatments of RNA virus infections. PLoS Pathog. 2009, 5, e1000658. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Domingo, E. Antiviral Strategies Based on Lethal Mutagenesis and Error Threshold. Curr. Top. Microbiol. Immunol. 2016, 392, 323–339. [Google Scholar] [PubMed]
- Kamar, N.; Rostaing, L.; Abravanel, F.; Garrouste, C.; Esposito, L.; Cardeau-Desangles, I.; Mansuy, J.M.; Selves, J.; Peron, J.M.; Otal, P.; et al. Pegylated interferon-alpha for treating chronic hepatitis E virus infection after liver transplantation. Clin. Infect. Dis. 2010, 50, e30–e33. [Google Scholar] [CrossRef] [PubMed]
- Haagsma, E.B.; Riezebos-Brilman, A.; van den Berg, A.P.; Porte, R.J.; Niesters, H.G. Treatment of chronic hepatitis E in liver transplant recipients with pegylated interferon alpha-2b. Liver Transpl. 2010, 16, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; Francois, C.; Anggakusuma; Behrendt, P.; Engelmann, M.; Knegendorf, L.; Vieyres, G.; Wedemeyer, H.; Hartmann, R.; Pietschmann, T.; et al. Antiviral Activities of Different Interferon Types and Subtypes against Hepatitis E Virus Replication. Antimicrob. Agents Chemother. 2016, 60, 2132–2139. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, L.; Wang, W.; Watashi, K.; Wang, Y.; Sprengers, D.; de Ruiter, P.E.; van der Laan, L.J.; Metselaar, H.J.; Kamar, N.; et al. Disparity of basal and therapeutically activated interferon signalling in constraining hepatitis E virus infection. J. Viral. Hepat. 2016, 23, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.P.; Jain, A.K.; Naik, G.; Soni, N.; Chitnis, D.S. Viral hepatitis during pregnancy. Int. J. Gynaecol. Obstet. 2001, 72, 103–108. [Google Scholar] [CrossRef]
- Khuroo, M.S.; Teli, M.R.; Skidmore, S.; Sofi, M.A.; Khuroo, M.I. Incidence and severity of viral hepatitis in pregnancy. Am. J. Med. 1981, 70, 252–255. [Google Scholar] [CrossRef]
- Behrendt, P.; Steinmann, E.; Manns, M.P.; Wedemeyer, H. The impact of hepatitis E in the liver transplant setting. J. Hepatol. 2014, 61, 1418–1429. [Google Scholar] [CrossRef] [PubMed]
- Pischke, S.; Suneetha, P.V.; Baechlein, C.; Barg-Hock, H.; Heim, A.; Kamar, N.; Schlue, J.; Strassburg, C.P.; Lehner, F.; Raupach, R.; et al. Hepatitis E virus infection as a cause of graft hepatitis in liver transplant recipients. Liver Transpl. 2010, 16, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Kamar, N.; Nicot, F.; Ducos, J.; Bismuth, M.; Garrigue, V.; Petitjean-Lecherbonnier, J.; Ollivier, I.; Alessandri-Gradt, E.; Goria, O.; et al. Mutation in the Hepatitis E Virus Polymerase and Outcome of Ribavirin Therapy. Antimicrob. Agents Chemother. 2016, 60, 1608–1614. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Emerson, S.U.; Wang, Y.; Pan, Q.; Balzarini, J.; Dallmeier, K.; Neyts, J. Ribavirin inhibits in vitro hepatitis E virus replication through depletion of cellular GTP pools and is moderately synergistic with alpha interferon. Antimicrob. Agents Chemother. 2014, 58, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, T.P.; Deeprose, R.D. Metabolism of 5-amino-1-beta-d-ribofuranosylimidazole-4-carboxamide and related five-membered heterocycles to 5′-triphosphates in human blood and L5178Y cells. Biochem. Pharmacol. 1978, 27, 709–716. [Google Scholar] [CrossRef]
- Muller, W.E.; Maidhof, A.; Taschner, H.; Zahn, R.K. Virazole (1-beta-d-ribofuranosyl-1,2,4-triazole-3-carboxamide; A cytostatic agent. Biochem. Pharmacol. 1977, 26, 1071–1075. [Google Scholar] [CrossRef]
- Varma, S.P.; Kumar, A.; Kapur, N.; Durgapal, H.; Acharya, S.K.; Panda, S.K. Hepatitis E virus replication involves alternating negative- and positive-sense RNA synthesis. J. Gen. Virol. 2011, 92 Pt 3, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Agudo, R.; Ferrer-Orta, C.; Arias, A.; de la Higuera, I.; Perales, C.; Perez-Luque, R.; Verdaguer, N.; Domingo, E. A multi-step process of viral adaptation to a mutagenic nucleoside analogue by modulation of transition types leads to extinction-escape. PLoS Pathog. 2010, 6, e1001072. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todt, D.; Walter, S.; Brown, R.J.P.; Steinmann, E. Mutagenic Effects of Ribavirin on Hepatitis E Virus—Viral Extinction versus Selection of Fitness-Enhancing Mutations. Viruses 2016, 8, 283. https://doi.org/10.3390/v8100283
Todt D, Walter S, Brown RJP, Steinmann E. Mutagenic Effects of Ribavirin on Hepatitis E Virus—Viral Extinction versus Selection of Fitness-Enhancing Mutations. Viruses. 2016; 8(10):283. https://doi.org/10.3390/v8100283
Chicago/Turabian StyleTodt, Daniel, Stephanie Walter, Richard J. P. Brown, and Eike Steinmann. 2016. "Mutagenic Effects of Ribavirin on Hepatitis E Virus—Viral Extinction versus Selection of Fitness-Enhancing Mutations" Viruses 8, no. 10: 283. https://doi.org/10.3390/v8100283
APA StyleTodt, D., Walter, S., Brown, R. J. P., & Steinmann, E. (2016). Mutagenic Effects of Ribavirin on Hepatitis E Virus—Viral Extinction versus Selection of Fitness-Enhancing Mutations. Viruses, 8(10), 283. https://doi.org/10.3390/v8100283