
HIV-1 Mutation and Recombination Rates Are Different in Macrophages and T-cells

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Molecular Clones
2.2. Cell Culture
2.3. Virus Production
2.4. Infections
2.5. Quantitative PCR
2.6. Amplifications for Sequencing
2.7. Sequence Alignment
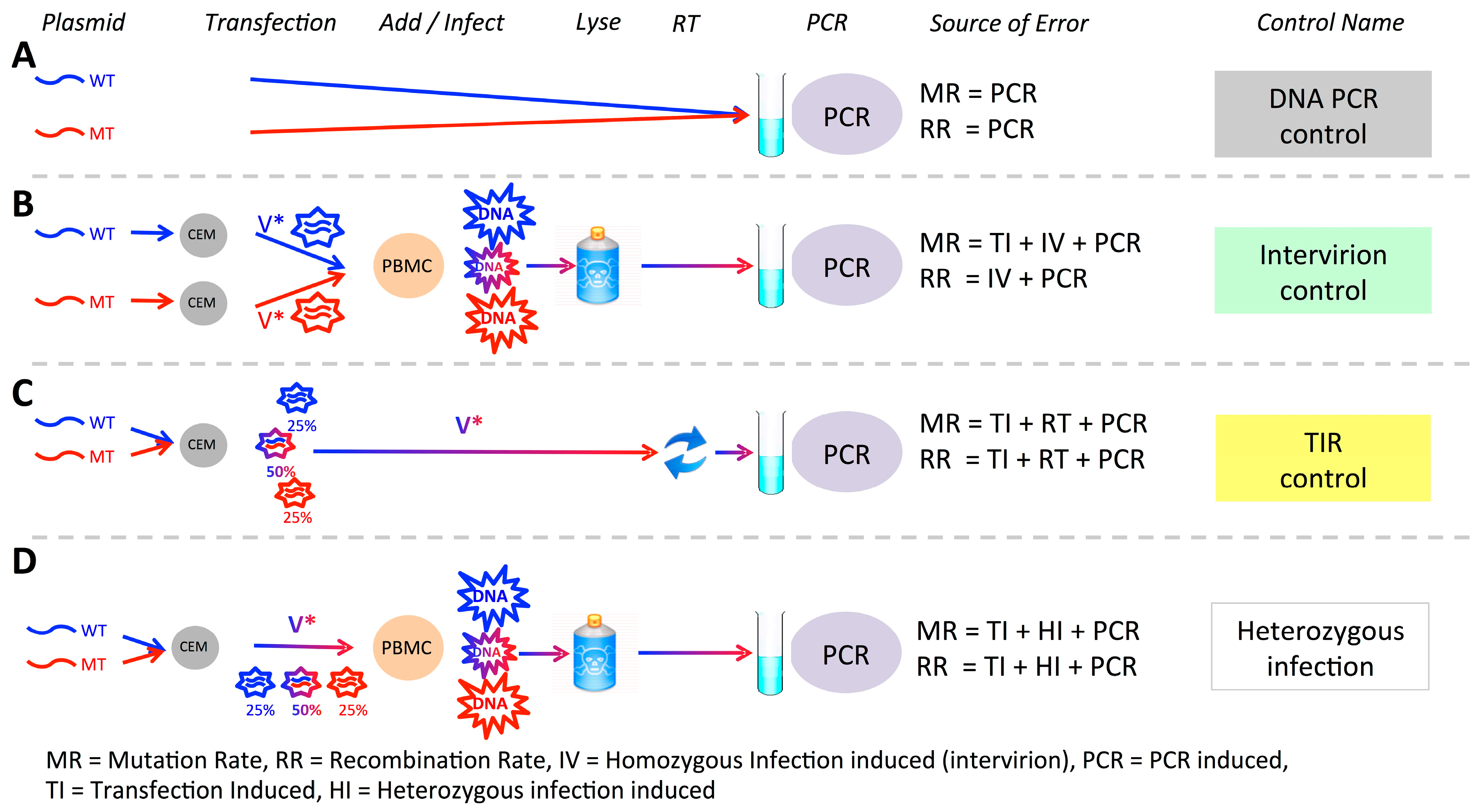
2.8. Controls
2.9. Mathematical Modeling
2.10. Statistical Methods
2.11. Association between Mutation and Recombination
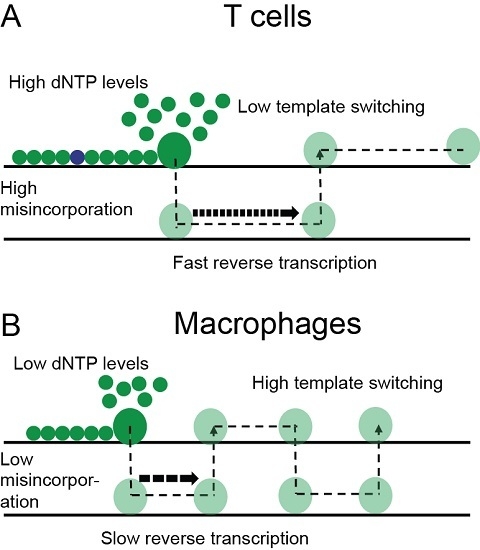
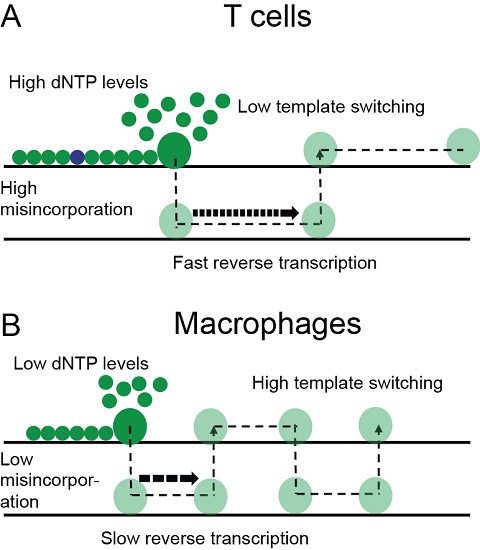
3. Results
3.1. Experimental System
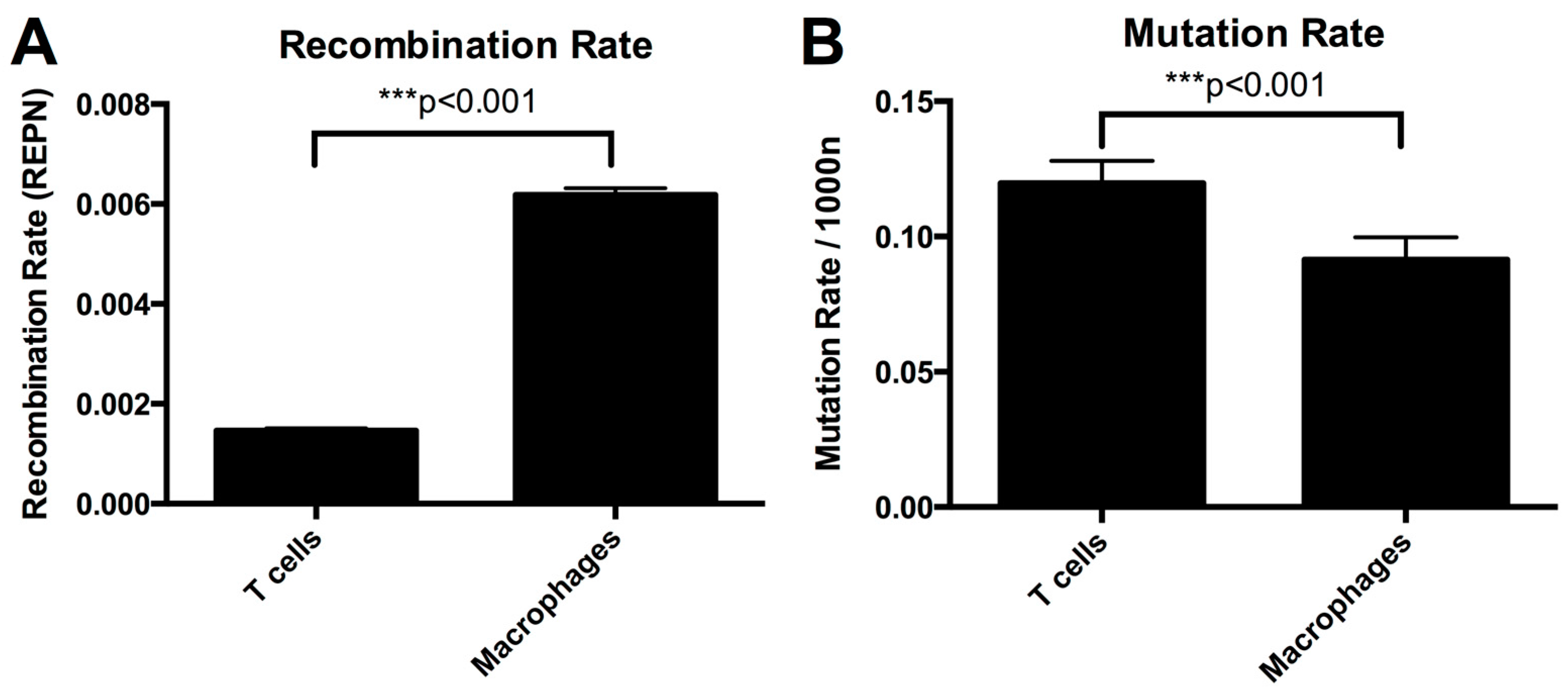
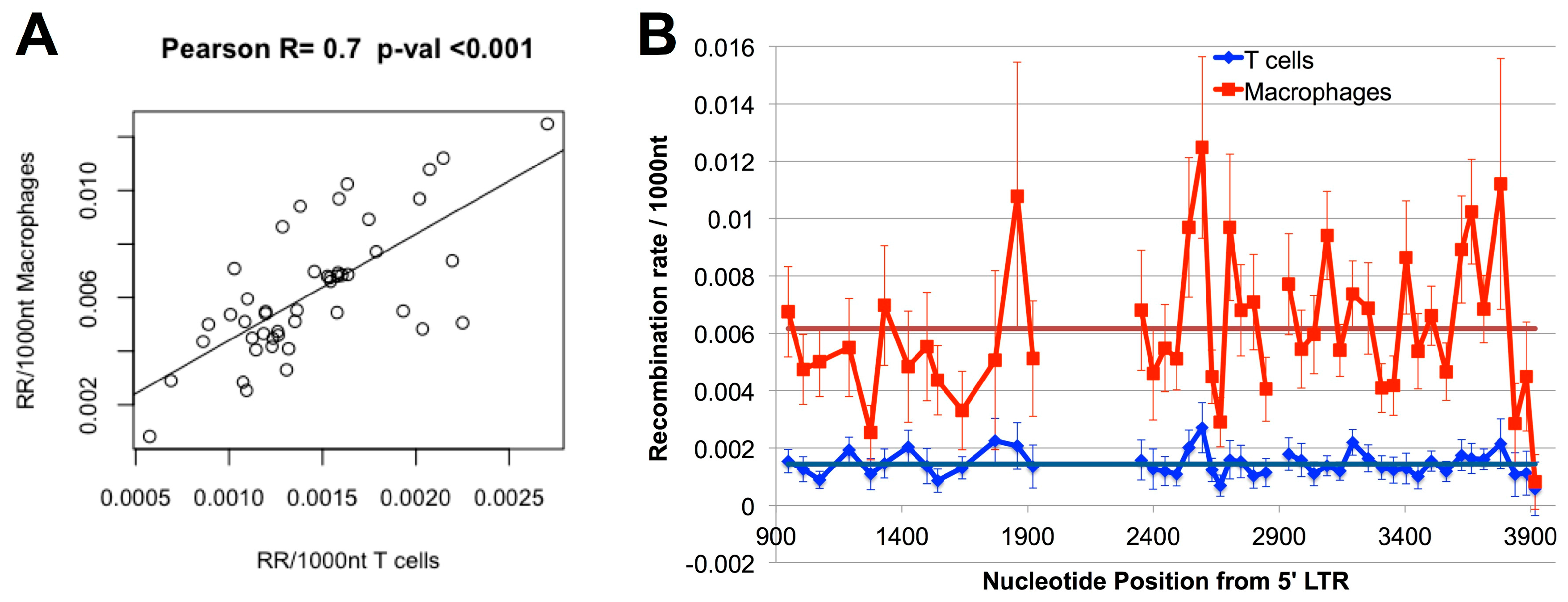
3.2. Higher Recombination Rate in Macrophages Compared to T-cells
3.3. The Difference in Recombination Rate between T-cells and Macrophages Is Not due to Means of Co-Receptor Entry or Sub-Population of T-cells
3.4. Hot and Cold Spots of Recombination Are Similar between T-cells and Macrophages
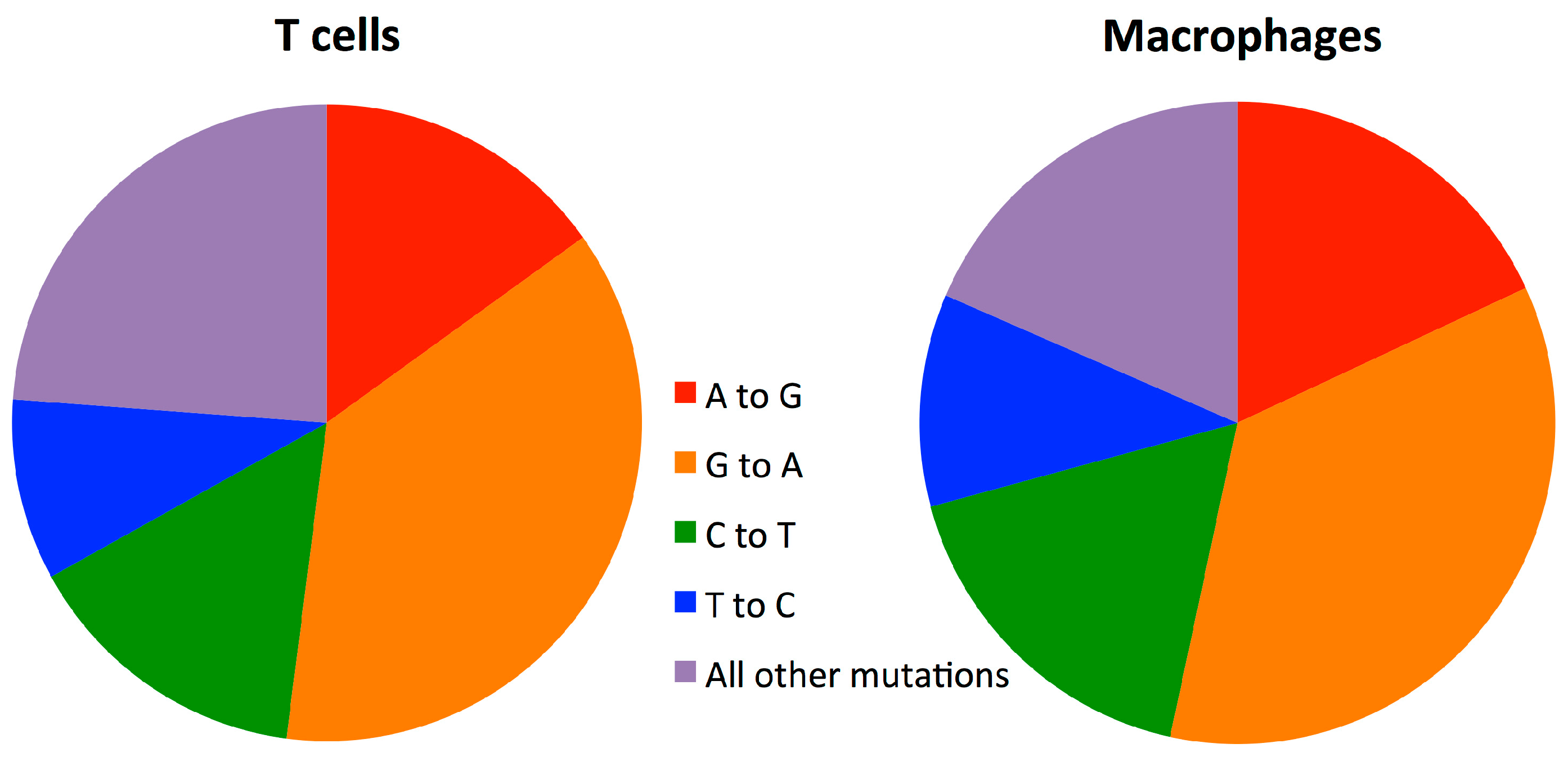
3.5. Mutation Rate in Macrophages Is Lower than in T-cells
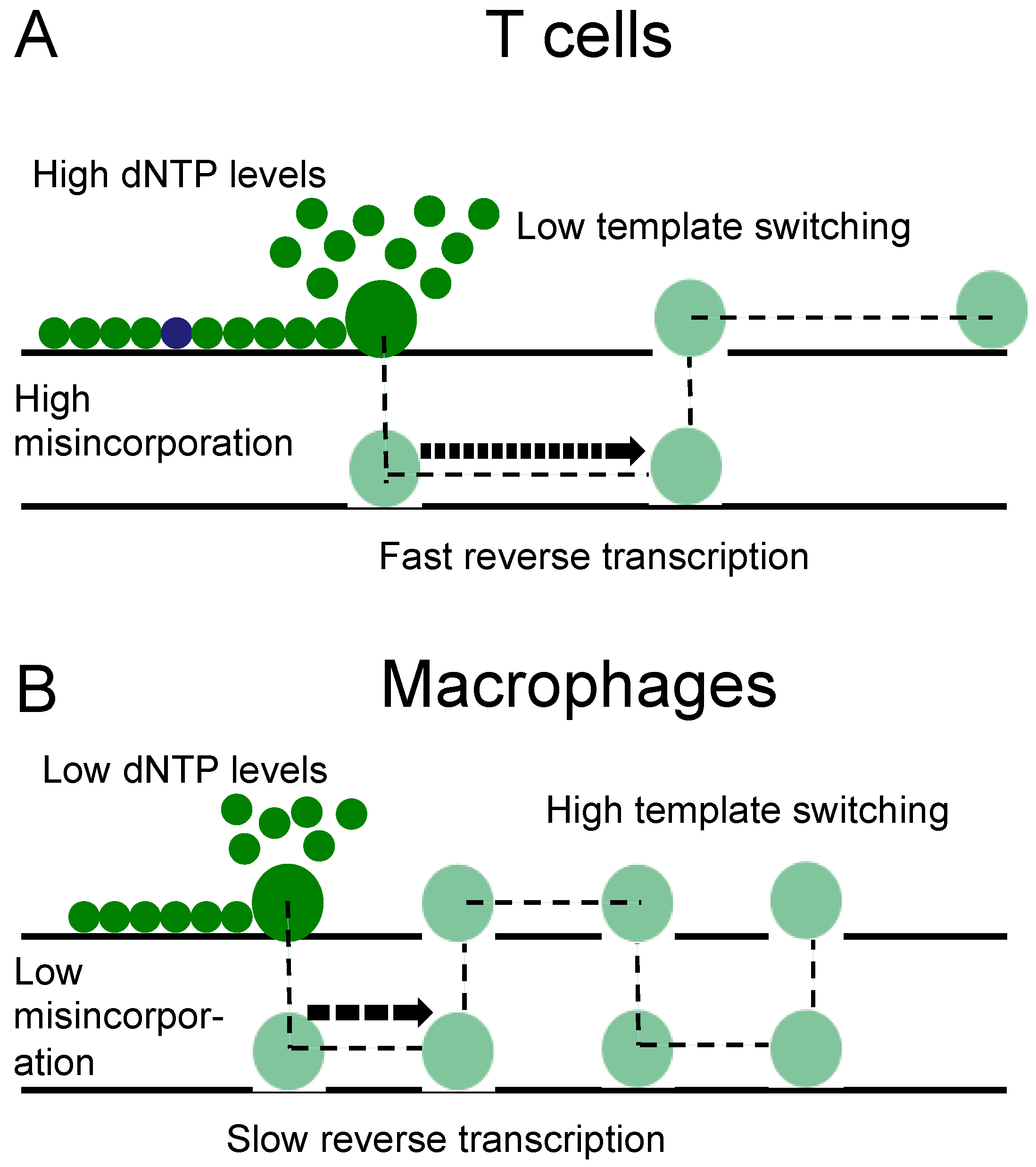
4. Discussion
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ho, D.; Neumann, A.; Perelson, A.; Chen, W.; Leonard, J.; Markowitz, M. Rapid turnover of plasma virions and cd4 lymphocytes in hiv-1 infection. Nature 1995, 373, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Smyth, R.P.; Davenport, M.P.; Mak, J. The origin of genetic diversity in hiv-1. Virus Res. 2012, 169, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Posada, D.; Crandall, K.A.; Holmes, E.C. The causes and consequences of hiv evolution. Nat. Rev. Genet. 2004, 5, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.S.; Stratov, I.; De Rose, R.; Walsh, K.; Dale, C.J.; Smith, M.Z.; Agy, M.B.; Hu, S.-L.; Krebs, K.; Watkins, D.I.; et al. Rapid viral escape at an immunodominant simian-human immunodeficiency virus cytotoxic t-lymphocyte epitope exacts a dramatic fitness cost. J. Virol. 2005, 79, 5721–5731. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.E.; Ferris, A.L.; Shao, W.; Alvord, W.G.; Hughes, S.H. Nature, position, and frequency of mutations made in a single cycle of hiv-1 replication. J. Virol. 2010, 84, 9864–9878. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M. Forward mutation rate of human immunodeficiency virus type 1 in a t lymphoid cell line. AIDS Res. Hum. Retrovir. 1996, 12, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [PubMed]
- Schlub, T.E.; Grimm, A.J.; Smyth, R.P.; Cromer, D.; Chopra, A.; Mallal, S.; Venturi, V.; Waugh, C.; Mak, J.; Davenport, M.P. 15%–20% of hiv substitution mutations are associated with recombination. J. Virol. 2014, 88, 3837–3849. [Google Scholar] [CrossRef] [PubMed]
- Holtz, C.M.; Mansky, L.M. Variation of hiv-1 mutation spectra among cell types. J. Virol. 2013, 87, 5296–5299. [Google Scholar] [CrossRef] [PubMed]
- Jetzt, A.E.; Yu, H.; Klarmann, G.J.; Ron, Y.; Preston, B.D.; Dougherty, J.P. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 2000, 74, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, T.; Wargo, H.; Hu, W.S. High rates of human immunodeficiency virus type 1 recombination: Near-random segregation of markers one kilobase apart in one round of viral replication. J. Virol. 2003, 77, 11193–11200. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Jetzt, A.E.; Sun, G.; Yu, H.; Klarmann, G.; Ron, Y.; Preston, B.D.; Dougherty, J.P. Human immunodeficiency virus type 1 recombination: Rate, fidelity, and putative hot spots. J. Virol. 2002, 76, 11273–11282. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rhodes, T.D.; Hu, W.-S. Comparison of the genetic recombination rates of human immunodeficiency virus type 1 in macrophages and t cells. J. Virol. 2005, 79, 9337–9340. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.N.; Aldrovandi, G.M.; Kutsch, O.; Shaw, G.M. Dynamics of hiv-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4204–4209. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.A.; Kim, D.-H.; Daly, M.B.; Allan, K.C.; Kim, B. Host samhd1 protein promotes hiv-1 recombination in macrophages. J. Biol. Chem. 2014, 289, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Schlub, T.; Smyth, R.; Grimm, A.; Mak, J.; Davenport, M.P. Accurately measuring recombination between closely related hiv-1 genomes. PLoS Comput. Biol. 2010, 6, e1000766. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits hiv-1 infection and is suppressed by the viral vif protein. Nat. 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Smyth, R.P.; Schlub, T.E.; Grimm, A.J.; Waugh, C.; Ellenberg, P.; Chopra, A.; Mallal, S.; Cromer, D.; Mak, J.; Davenport, M.P. Identifying recombination hot spots in the hiv-1 genome. J. Virol. 2014, 88, 2891–2902. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.S.; Regier, D.A.; Desrosiers, R.C. Construction and in vitro properties of hiv-1 mutants with deletions in “nonessential” genes. AIDS Res. Hum. Retrovir. 1994, 10, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S. Hiv-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell 1990, 61, 213–222. [Google Scholar] [CrossRef]
- Smyth, R.P.; Schlub, T.E.; Grimm, A.; Venturi, V.; Chopra, A.; Mallal, S.; Davenport, M.P.; Mak, J. Reducing chimera formation during pcr amplification to ensure accurate genotyping. Gene 2010, 469, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Stenzel, U.; Myles, S.; Prufer, K.; Hofreiter, M. Targeted high-throughput sequencing of tagged nucleic acid samples. Nucleic Acids Res. 2007, 35, e97. [Google Scholar] [CrossRef] [PubMed]
- Waugh, C.; Cromer, D.; Grimm, A.; Chopra, A.; Mallal, S.; Davenport, M.; Mak, J. A general method to eliminate laboratory induced recombinants during massive, parallel sequencing of cdna library. Virol. J. 2015, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Huse, S.M.; Huber, J.A.; Morrison, H.G.; Sogin, M.L.; Welch, D.M. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007, 8, R143. [Google Scholar] [CrossRef] [PubMed]
- Goto, N.; Prins, P.; Nakao, M.; Bonnal, R.; Aerts, J.; Katayama, T. Bioruby: Bioinformatics software for the ruby programming language. Bioinformatics (Oxf. Engl.) 2010, 26, 2617–2619. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2011. [Google Scholar]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.-G. Statistical power analyses using g*power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Clapham, P.R.; McKnight, A. Hiv-1 receptors and cell tropism. Br. Med.l Bull. 2001, 58, 43–59. [Google Scholar] [CrossRef]
- Rawson, J.M.O.; Landman, S.R.; Reilly, C.S.; Mansky, L.M. Hiv-1 and hiv-2 exhibit similar mutation frequencies and spectra in the absence of g-to-a hypermutation. Retrovirology 2015, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Daddacha, W.; Noble, E.; Nguyen, L.A.; Kennedy, E.M.; Kim, B. Effect of ribonucleotides embedded in a DNA template on hiv-1 reverse transcription kinetics and fidelity. J. Biol. Chem. 2013, 288, 12522–12532. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.K.; Chen, R.; Skasko, M.; Reynolds, H.M.; Lee, K.; Bambara, R.A.; Mansky, L.M.; Kim, B. A role for dntp binding of human immunodeficiency virus type 1 reverse transcriptase in viral mutagenesis. Biochemistry 2004, 43, 4490–4500. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Hanson, M.N.; Balakrishnan, M.; Boyer, P.L.; Roques, B.P.; Hughes, S.H.; Kim, B.; Bambara, R.A. Apparent defects in processive DNA synthesis, strand transfer, and primer elongation of met-184 mutants of hiv-1 reverse transcriptase derive solely from a dntp utilization defect. J. Biol. Chem. 2008, 283, 9196–9205. [Google Scholar] [CrossRef] [PubMed]
- Operario, D.J.; Balakrishnan, M.; Bambara, R.A.; Kim, B. Reduced dntp interaction of human immunodeficiency virus type 1 reverse transcriptase promotes strand transfer. J. Biol. Chem. 2006, 281, 32113–32121. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Type | # Nucleotides | MR/1000nt | RR/1000nt * |
|---|---|---|---|
| PCR | 4,931,024 | 0.074 | 0.015 |
| Intervirion (T-cells) | 15,407,933 | 0.119 | 0.044 |
| Intervirion (macrophages) | 10,947,705 | 0.097 | 0.047 |
| Cell Type | # Nucleotides | # Recombs | # Mutations | MR/1000nt | RR/1000nt * |
|---|---|---|---|---|---|
| T-cells | 7,180,718 | 4801 | 859 | 0.120 | 1.465 |
| Macrophages | 5,731,387 | 12,769 | 524 | 0.091 | 6.184 |
| Donor | RR/1000nt T-cells | RR/1000nt Macrophages | MR/1000nt T-cells | MR/1000nt Macrophages |
|---|---|---|---|---|
| Donor A | 1.73 | 3.82 | 0.120 | 0.092 |
| Donor B | 1.13 | 7.25 | 0.118 | 0.085 |
| Donor C | 1.55 | 7.61 | 0.121 | 0.098 |
| T-cells | Macrophages | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Orig Nucl | # Nucl | % of Muts | % Mutating to | # Nucl | % of Muts | % Mutating to | ||||||
| A | C | G | T | A | C | G | T | |||||
| A | 2,745,580 | 20.6 | 0.0 | 3.0 | 15.1 | 2.4 | 2,275,211 | 22.2 | 0.0 | 1.1 | 18.1 | 3.0 |
| C | 1,300,503 | 22.8 | 6.8 | 0.0 | 1.2 | 14.9 | 1,069,497 | 24.2 | 5.5 | 0.0 | 1.3 | 17.4 |
| G | 1,576,904 | 42.4 | 36.9 | 1.0 | 0.0 | 4.4 | 1,297,412 | 40.5 | 35.3 | 1.1 | 0.0 | 4.1 |
| T | 1,557,725 | 14.2 | 2.2 | 9.2 | 2.8 | 0.0 | 1,293,501 | 13.1 | 0.2 | 10.7 | 2.2 | 0.0 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cromer, D.; Schlub, T.E.; Smyth, R.P.; Grimm, A.J.; Chopra, A.; Mallal, S.; Davenport, M.P.; Mak, J. HIV-1 Mutation and Recombination Rates Are Different in Macrophages and T-cells. Viruses 2016, 8, 118. https://doi.org/10.3390/v8040118
Cromer D, Schlub TE, Smyth RP, Grimm AJ, Chopra A, Mallal S, Davenport MP, Mak J. HIV-1 Mutation and Recombination Rates Are Different in Macrophages and T-cells. Viruses. 2016; 8(4):118. https://doi.org/10.3390/v8040118
Chicago/Turabian StyleCromer, Deborah, Timothy E. Schlub, Redmond P. Smyth, Andrew J. Grimm, Abha Chopra, Simon Mallal, Miles P. Davenport, and Johnson Mak. 2016. "HIV-1 Mutation and Recombination Rates Are Different in Macrophages and T-cells" Viruses 8, no. 4: 118. https://doi.org/10.3390/v8040118
APA StyleCromer, D., Schlub, T. E., Smyth, R. P., Grimm, A. J., Chopra, A., Mallal, S., Davenport, M. P., & Mak, J. (2016). HIV-1 Mutation and Recombination Rates Are Different in Macrophages and T-cells. Viruses, 8(4), 118. https://doi.org/10.3390/v8040118