Polyamidoamine Nanoparticles for the Oral Administration of Antimalarial Drugs

 , ,
, ,  ,
, 


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Polyamidoamine (PAA) Synthesis and Characterization
2.2. Matrix-Assisted Laser Desorption/Ionization Tandem Time-of-Flight (MALDI TOF/TOF) Analysis
2.3. Plasmodium falciparum Cell Culture and Parasite Growth Inhibition, Hemolysis and Unspecific Cytotoxicity Assays
2.4. Cell Targeting Analysis
2.5. Generation of Polyclonal Antibodies against PAAs
2.6. Affinity Chromatography
2.7. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
2.8. Transmission Electron Microscopy (TEM) of Cell Sections
2.9. Determination of PAA-FITC Presence in Blood and Toxicity Assays in Mice
2.10. Antimalarial Activity Assay In Vivo
2.11. PAA Administration to Mosquitoes
2.12. Statistical Analysis
3. Results
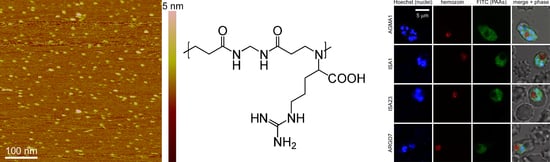
3.1. Characterization of PAAs and Stability Analysis in GIT Conditions
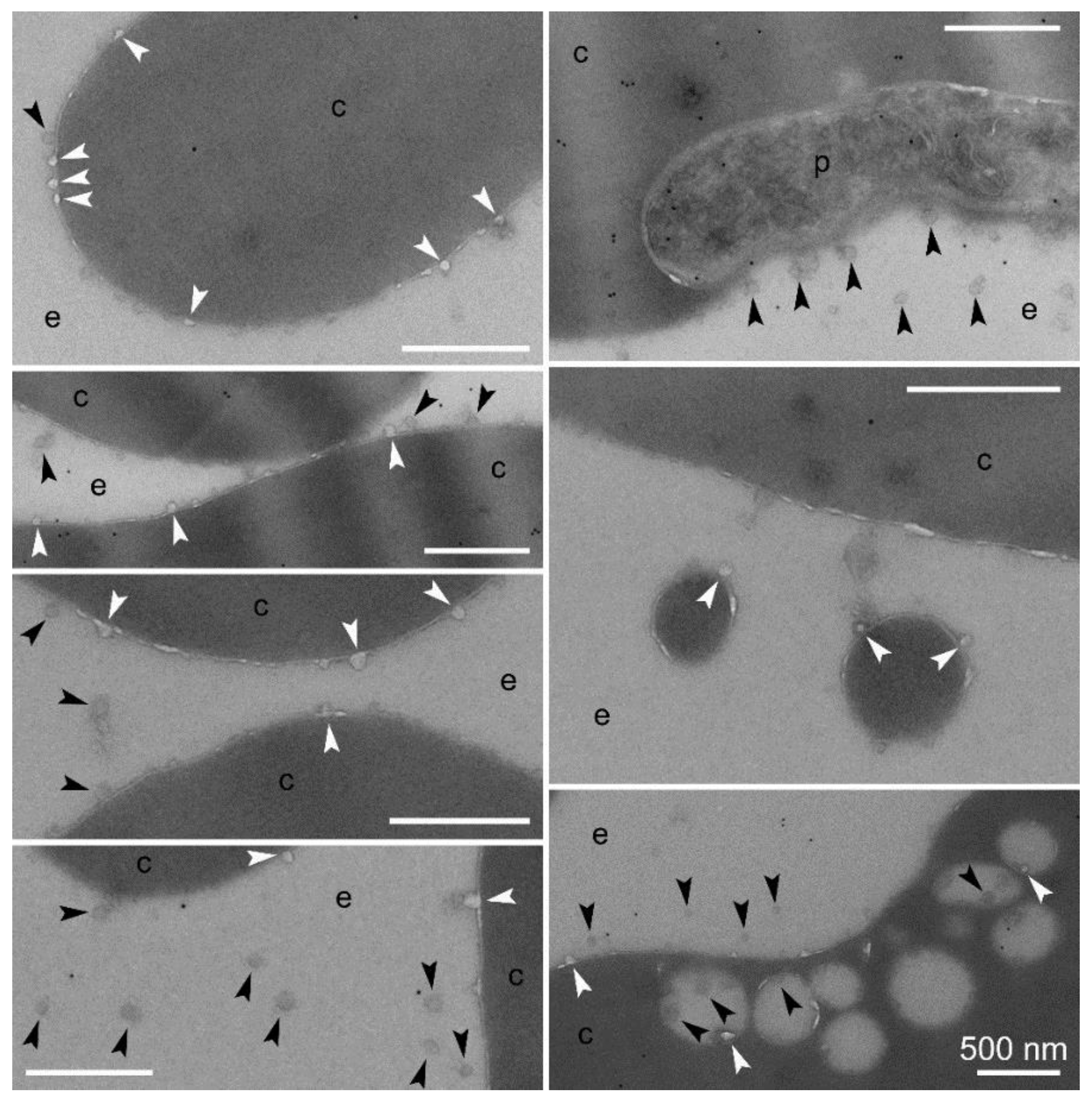
3.2. Cell Targeting of PAAs
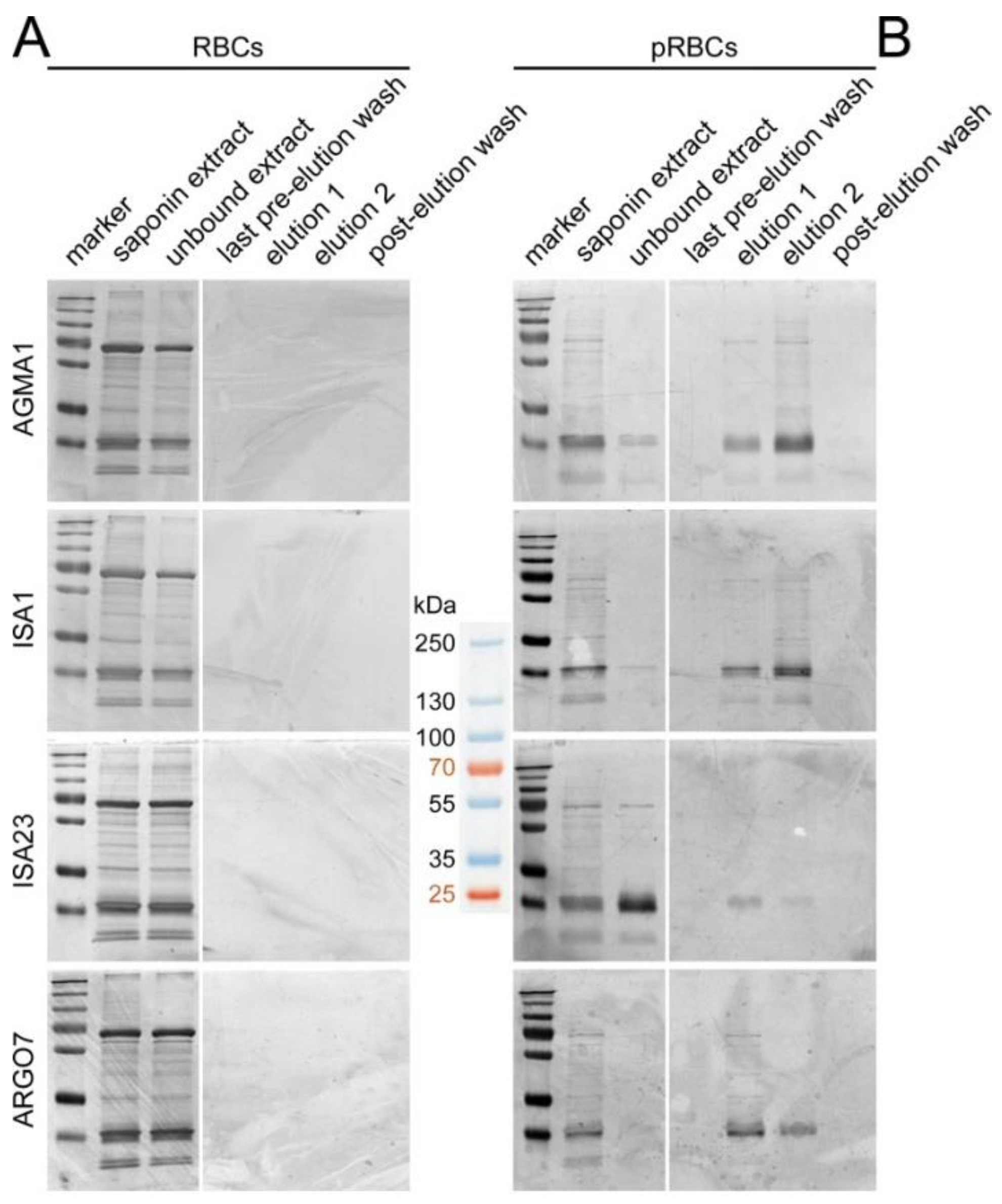
3.3. Affinity Chromatography Analysis of PAA-Binding pRBC Proteins
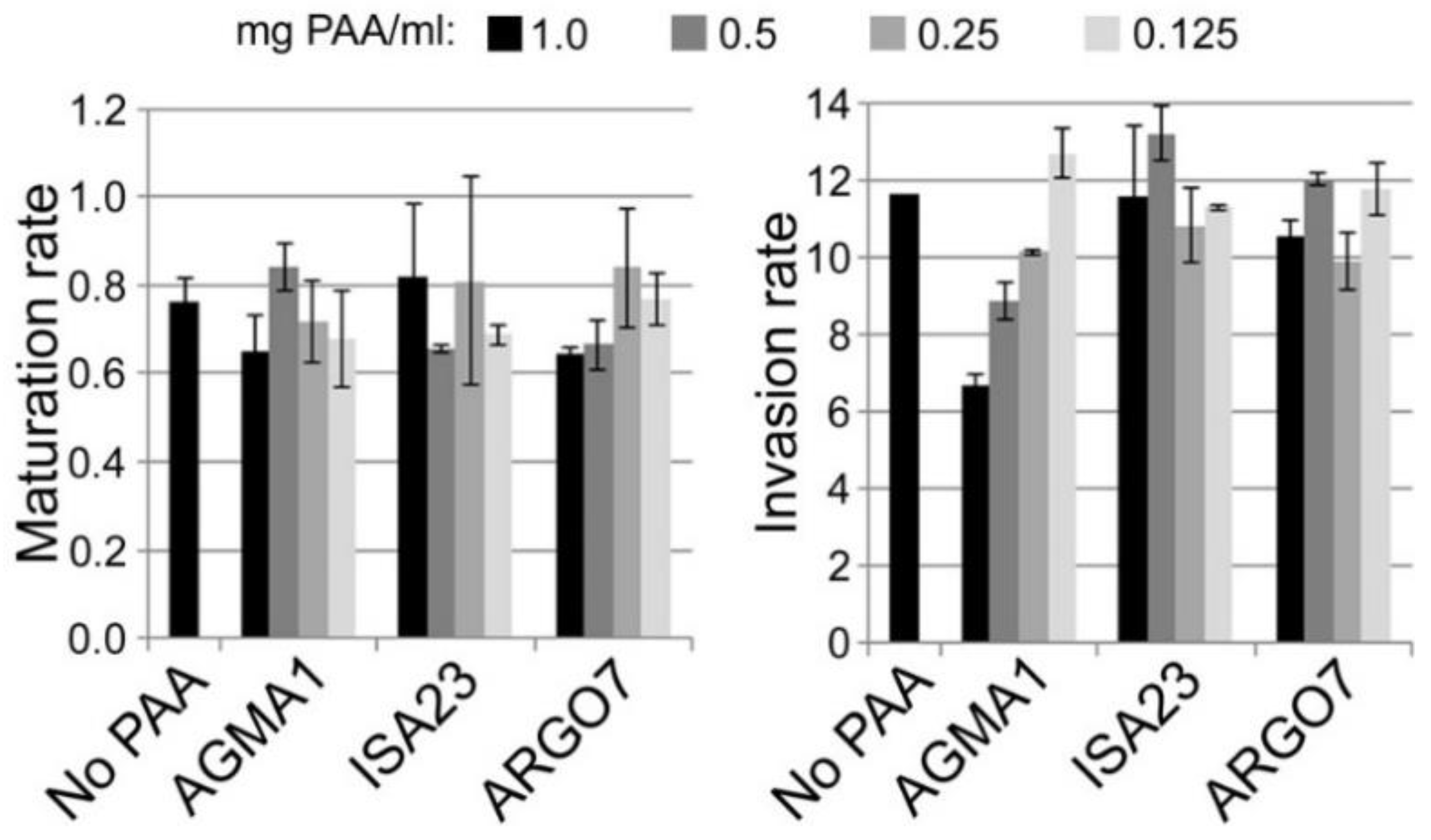
3.4. In Vitro Plasmodium Growth Inhibition Assays
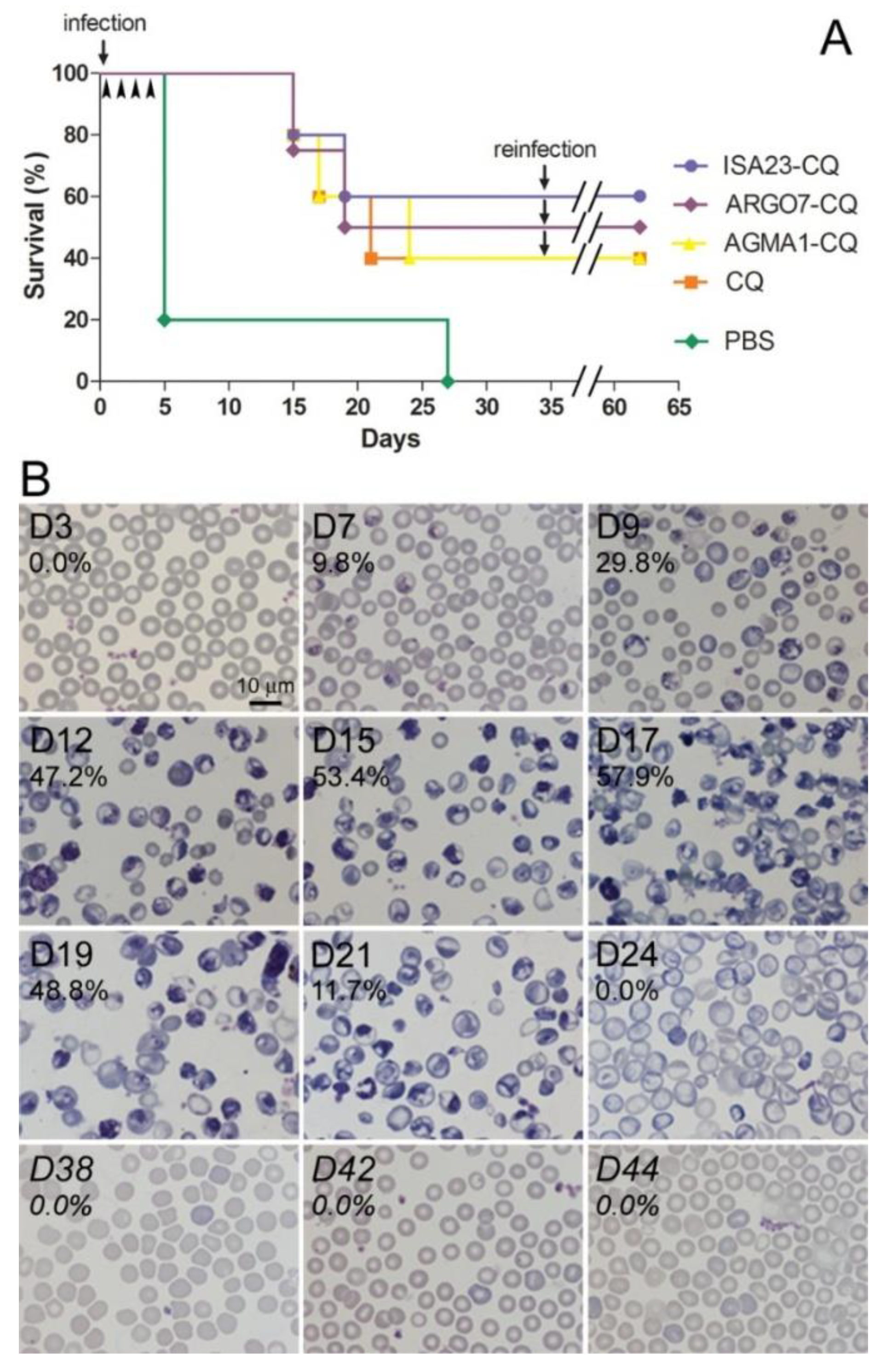
3.5. Oral Administration to Mice
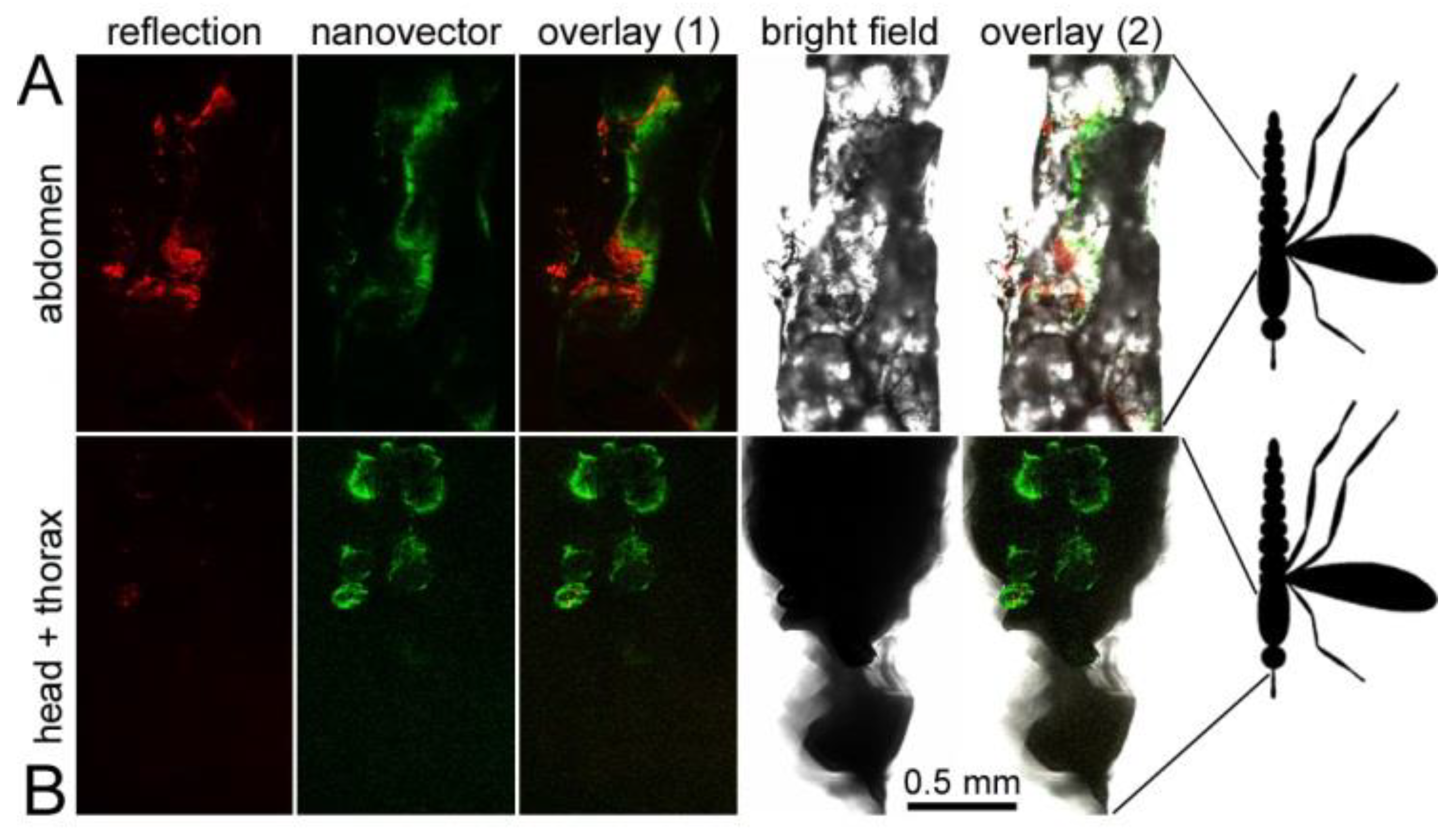
3.6. Oral Administration to Mosquitoes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015; Available online: http://apps.who.int/iris/bitstream/10665/162441/1/9789241549127_eng.pdf (accessed on 13 July 2018).
- Bergström, C.A.S.; Holm, R.; Jørgensen, S.A.; Andersson, S.B.E.; Artursson, P.; Beato, S.; Borde, A.; Box, K.; Brewster, M.; Dressman, J.; et al. Early pharmaceutical profiling to predict oral drug absorption: Current status and unmet needs. Eur. J. Pharm. Sci. 2014, 57 (Suppl. C), 173–199. [Google Scholar] [CrossRef] [PubMed]
- Plapied, L.; Duhem, N.; des Rieux, A.; Preat, V. Fate of polymeric nanocarriers for oral drug delivery. Curr. Opin. Colloid Interface Sci. 2011, 16, 228–237. [Google Scholar] [CrossRef]
- Ryan, K.B.; Maher, S.; Brayden, D.J.; O’Driscoll, C.M. Nanostructures overcoming the intestinal barrier: Drug delivery strategies. In Nanostructured Biomaterials for Overcoming Biological Barriers, 1st ed.; Alonso, M.J., Csaba, N.S., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2012; pp. 63–90. ISBN 978-1-84973-363-2. [Google Scholar]
- Raemdonck, K.; Braeckmans, K.; Demeester, J.; De Smedt, S.C. Merging the best of both worlds: Hybrid lipid-enveloped matrix nanocomposites in drug delivery. Chem. Soc. Rev. 2014, 43, 444–472. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.D.; Gauthier, M.A.; Leroux, J.C. Improving oral drug bioavailability with polycations? Eur. J. Pharm. Biopharm. 2015, 97 Pt B, 427–437. [Google Scholar] [CrossRef]
- Sweet, D.M.; Kolhatkar, R.B.; Ray, A.; Swaan, P.; Ghandehari, H. Transepithelial transport of PEGylated anionic poly(amidoamine) dendrimers: Implications for oral drug delivery. J. Control. Release 2009, 138, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellepeddi, V.K.; Ghandehari, H. Poly(amido amine) dendrimers in oral delivery. Tissue Barriers 2016, 4, e1173773. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, G.; Greish, K.; Ghandehari, H. Charge affects the oral toxicity of poly(amido amine) dendrimers. Eur. J. Pharm. Biopharm. 2013, 84, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, Z.; Gao, W.; Chen, Q.; Yu, B. Polyamidoamine dendrimers as potential drug carriers for enhanced aqueous solubility and oral bioavailability of silybin. Drug Dev. Ind. Pharm. 2011, 37, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Urbán, P.; Valle-Delgado, J.J.; Mauro, N.; Marques, J.; Manfredi, A.; Rottmann, M.; Ranucci, E.; Ferruti, P.; Fernàndez-Busquets, X. Use of poly(amidoamine) drug conjugates for the delivery of antimalarials to Plasmodium. J. Control. Release 2014, 177, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Urbán, P.; Ranucci, E.; Fernàndez-Busquets, X. Polyamidoamine nanoparticles as nanocarriers for the drug delivery to malaria parasite stages in the mosquito vector. Nanomedicine 2015, 10, 3401–3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranucci, E.; Ferruti, P.; Lattanzio, E.; Manfredi, A.; Rossi, M.; Mussini, P.R.; Chiellini, F.; Bartoli, C. Acid-base properties of poly(amidoamine)s. J. Polym. Sci. A Polym. Chem. 2009, 47, 6977–6991. [Google Scholar] [CrossRef]
- Lavignac, N.; Nicholls, J.L.; Ferruti, P.; Duncan, R. Poly(amidoamine) conjugates containing doxorubicin bound via an acid-sensitive linker. Macromol. Biosci. 2009, 9, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferruti, P.; Franchini, J.; Bencini, M.; Ranucci, E.; Zara, G.P.; Serpe, L.; Primo, L.; Cavalli, R. Prevailingly cationic agmatine-based amphoteric polyamidoamine as a nontoxic, nonhemolytic, and “stealthlike” DNA complexing agent and transfection promoter. Biomacromolecules 2007, 8, 1498–1504. [Google Scholar] [CrossRef] [PubMed]
- Ferruti, P.; Mauro, N.; Falciola, L.; Pifferi, V.; Bartoli, C.; Gazzarri, M.; Chiellini, F.; Ranucci, E. Amphoteric, prevailingly cationic l-arginine polymers of poly(amidoamino acid) structure: Synthesis, acid/base properties and preliminary cytocompatibility and cell-permeating characterizations. Macromol. Biosci. 2014, 14, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Ranucci, E.; Spagnoli, G.; Latini, R.; Bernasconi, R.; Ferruti, P. On the suitability of urethane bonds between the carrier and the drug moiety in poly(ethyleneglycol)-based oligomeric prodrugs. J. Biomater. Sci. Polym. Ed. 1994, 6, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Donghi, D.; Maggioni, D.; D’Alfonso, G.; Amigoni, F.; Ranucci, E.; Ferruti, P.; Manfredi, A.; Fenili, F.; Bisazza, A.; Cavalli, R. Tricarbonyl-rhenium complexes of a thiol-functionalized amphoteric poly(amidoamine). Biomacromolecules 2009, 10, 3273–3282. [Google Scholar] [CrossRef] [PubMed]
- Ferruti, P.; Ranucci, E.; Trotta, F.; Gianasi, E.; Evagorou, E.G.; Wasil, M.; Wilson, G.; Duncan, R. Synthesis, characterisation and antitumour activity of platinum(II) complexes of novel functionalised poly(amido amine)s. Macromol. Chem. Phys. 1999, 200, 1644–1654. [Google Scholar] [CrossRef]
- Urbán, P.; Estelrich, J.; Cortés, A.; Fernàndez-Busquets, X. A nanovector with complete discrimination for targeted delivery to Plasmodium falciparum-infected versus non-infected red blood cells in vitro. J. Control. Release 2011, 151, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Cranmer, S.L.; Magowan, C.; Liang, J.; Coppel, R.L.; Cooke, B.M. An alternative to serum for cultivation of Plasmodium falciparum in vitro. Trans. R. Soc. Trop. Med. Hyg. 1997, 91, 363–365. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.; Vilanova, E.; Mourão, P.A.S.; Fernàndez-Busquets, X. Marine organism sulfated polysaccharides exhibiting significant antimalarial activity and inhibition of red blood cell invasion by Plasmodium. Sci. Rep. 2016, 6, 24368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council (US). Guide for the Care and Use of Laboratory Animals, 8th ed.; The National Academies Press: Washington, DC, USA, 2011; ISBN 978-0-309-15400-0. [Google Scholar]
- Moles, E.; Urbán, P.; Jiménez-Díaz, M.B.; Viera-Morilla, S.; Angulo-Barturen, I.; Busquets, M.A.; Fernàndez-Busquets, X. Immunoliposome-mediated drug delivery to Plasmodium-infected and non-infected red blood cells as a dual therapeutic/prophylactic antimalarial strategy. J. Control. Release 2015, 210, 217–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Switzer, R.C., III; Merril, C.R.; Shifrin, S. A highly sensitive silver stain for detecting proteins and peptides in polyacrylamide gels. Anal. Biochem. 1979, 98, 231–237. [Google Scholar] [CrossRef]
- Schliwa, M.; van Blerkom, J. Structural interaction of cytoskeletal components. J. Cell Biol. 1981, 90, 222–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidock, D.A.; Rosenthal, P.J.; Croft, S.L.; Brun, R.; Nwaka, S. Antimalarial drug discovery: Efficacy models for compound screening. Nat. Rev. Drug Discov. 2004, 3, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Ferruti, P.; Marchisio, M.A.; Duncan, R. Polyamidoamines: Biomedical applications. Macromol. Rapid Commun. 2002, 23, 332–355. [Google Scholar] [CrossRef]
- Ofulla, A.O.; Orago, A.S.; Githure, J.I.; Burans, J.P.; Aleman, G.M.; Johnson, A.J.; Martin, S.K. Determination of fifty percent inhibitory concentrations (IC50) of antimalarial drugs against Plasmodium falciparum parasites in a serum-free medium. Am. J. Trop. Med. Hyg. 1994, 51, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Regev-Rudzki, N.; Wilson, D.; Carvalho, T.; Sisquella, X.; Coleman, B.; Rug, M.; Bursac, D.; Angrisano, F.; Gee, M.; Hill, A.; et al. Cell-cell communication between malaria-infected red blood cells via exosome-like vesicles. Cell 2013, 153, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Yang, C.; Patterson, P.S.; Udhayakumar, V.; Lal, A.A. Sulfated polyanions inhibit invasion of erythrocytes by plasmodial merozoites and cytoadherence of endothelial cells to parasitized erythrocytes. Infect. Immun. 1996, 64, 1373–1378. [Google Scholar] [PubMed]
- Najer, A.; Wu, D.; Bieri, A.; Brand, F.; Palivan, C.G.; Beck, H.P.; Meier, W. Nanomimics of host cell membranes block invasion and expose invasive malaria parasites. ACS Nano 2014, 8, 12560–12571. [Google Scholar] [CrossRef] [PubMed]
- Bastos, M.F.; Albrecht, L.; Kozlowski, E.O.; Lopes, S.C.P.; Blanco, Y.C.; Carlos, B.C.; Castiñeiras, C.; Vicente, C.P.; Werneck, C.C.; Wunderlich, G.; et al. Fucosylated chondroitin sulfate inhibits Plasmodium falciparum cytoadhesion and merozoite invasion. Antimicrob. Agents Chemother. 2014, 58, 1862–1871. [Google Scholar] [CrossRef] [PubMed]
- Kulane, A.; Ekre, H.P.; Perlmann, P.; Rombo, L.; Wahlgren, M.; Wahlin, B. Effect of different fractions of heparin on Plasmodium falciparum merozoite invasion of red blood cells in vitro. Am. J. Trop. Med. Hyg. 1992, 46, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.J.; Richards, J.S.; Gilson, P.R.; Chai, W.; Beeson, J.G. Interactions with heparin-like molecules during erythrocyte invasion by Plasmodium falciparum merozoites. Blood 2010, 115, 4559–4568. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, J.A.; Thomas, A.W.; Hall, T.; Chulay, J.D. Specificities of antibodies that inhibit merozoite dispersal from malaria-infected erythrocytes. Mol. Biochem. Parasitol. 1989, 36, 77–85. [Google Scholar] [CrossRef]
- Pasvol, G. Receptors on red cells for Plasmodium falciparum and their interaction with merozoites. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1984, 307, 189. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K. Membrane transport in the malaria-infected erythrocyte. Physiol. Rev. 2001, 81, 495–537. [Google Scholar] [CrossRef] [PubMed]
- Goodyer, I.D.; Pouvelle, B.; Schneider, T.G.; Trelka, D.P.; Taraschi, T.F. Characterization of macromolecular transport pathways in malaria-infected erythrocytes. Mol. Biochem. Parasitol. 1997, 87, 13–28. [Google Scholar] [CrossRef]
- Marques, J.; Moles, E.; Urbán, P.; Prohens, R.; Busquets, M.A.; Sevrin, C.; Grandfils, C.; Fernàndez-Busquets, X. Application of heparin as a dual agent with antimalarial and liposome targeting activities towards Plasmodium-infected red blood cells. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.; Valle-Delgado, J.J.; Urbán, P.; Baró, E.; Prohens, R.; Mayor, A.; Cisteró, P.; Delves, M.; Sinden, R.E.; Grandfils, C.; et al. Adaptation of targeted nanocarriers to changing requirements in antimalarial drug delivery. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Gulati, N.M.; Stewart, P.L.; Steinmetz, N.F. Bioinspired shielding strategies for nanoparticle drug delivery applications. Mol. Pharm. 2018, 15, 2900–2909. [Google Scholar] [CrossRef] [PubMed]
- Paaijmans, K.; Fernàndez-Busquets, X. Antimalarial drug delivery to the mosquito: An option worth exploring? Future Microbiol. 2014, 9, 579–582. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martí Coma-Cros, E.; Biosca, A.; Marques, J.; Carol, L.; Urbán, P.; Berenguer, D.; Riera, M.C.; Delves, M.; Sinden, R.E.; Valle-Delgado, J.J.; et al. Polyamidoamine Nanoparticles for the Oral Administration of Antimalarial Drugs. Pharmaceutics 2018, 10, 225. https://doi.org/10.3390/pharmaceutics10040225
Martí Coma-Cros E, Biosca A, Marques J, Carol L, Urbán P, Berenguer D, Riera MC, Delves M, Sinden RE, Valle-Delgado JJ, et al. Polyamidoamine Nanoparticles for the Oral Administration of Antimalarial Drugs. Pharmaceutics. 2018; 10(4):225. https://doi.org/10.3390/pharmaceutics10040225
Chicago/Turabian StyleMartí Coma-Cros, Elisabet, Arnau Biosca, Joana Marques, Laura Carol, Patricia Urbán, Diana Berenguer, Maria Cristina Riera, Michael Delves, Robert E. Sinden, Juan José Valle-Delgado, and et al. 2018. "Polyamidoamine Nanoparticles for the Oral Administration of Antimalarial Drugs" Pharmaceutics 10, no. 4: 225. https://doi.org/10.3390/pharmaceutics10040225
APA StyleMartí Coma-Cros, E., Biosca, A., Marques, J., Carol, L., Urbán, P., Berenguer, D., Riera, M. C., Delves, M., Sinden, R. E., Valle-Delgado, J. J., Spanos, L., Siden-Kiamos, I., Pérez, P., Paaijmans, K., Rottmann, M., Manfredi, A., Ferruti, P., Ranucci, E., & Fernàndez-Busquets, X. (2018). Polyamidoamine Nanoparticles for the Oral Administration of Antimalarial Drugs. Pharmaceutics, 10(4), 225. https://doi.org/10.3390/pharmaceutics10040225